 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraphPINE: Graph Importance Propagation for Interpretable Drug Response Prediction
Apr 07, 2025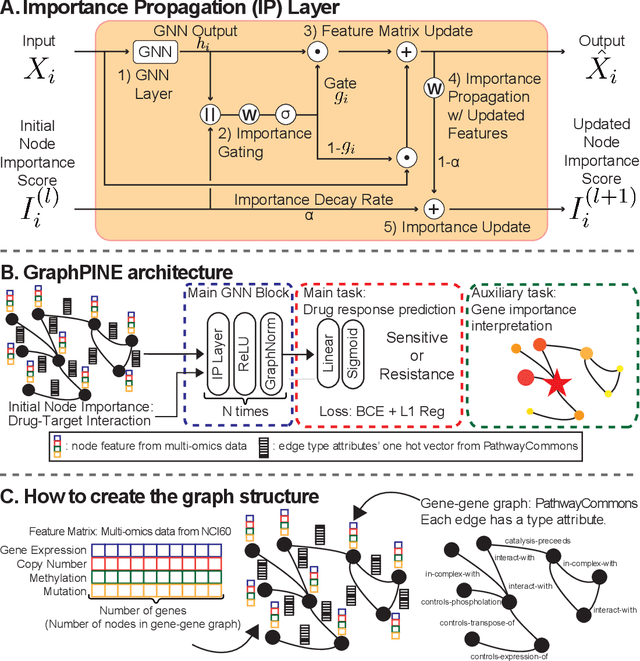

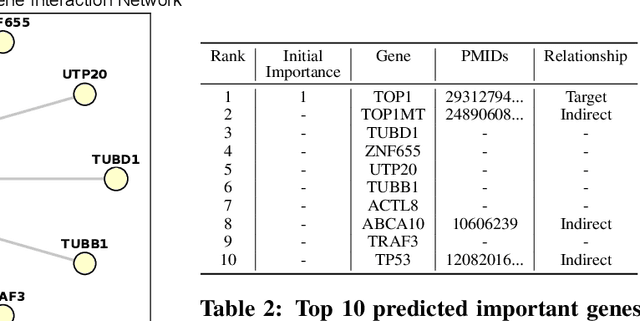

Explainability is necessary for many tasks in biomedical research. Recent explainability methods have focused on attention, gradient, and Shapley value. These do not handle data with strong associated prior knowledge and fail to constrain explainability results based on known relationships between predictive features. We propose GraphPINE, a graph neural network (GNN) architecture leveraging domain-specific prior knowledge to initialize node importance optimized during training for drug response prediction. Typically, a manual post-prediction step examines literature (i.e., prior knowledge) to understand returned predictive features. While node importance can be obtained for gradient and attention after prediction, node importance from these methods lacks complementary prior knowledge; GraphPINE seeks to overcome this limitation. GraphPINE differs from other GNN gating methods by utilizing an LSTM-like sequential format. We introduce an importance propagation layer that unifies 1) updates for feature matrix and node importance and 2) uses GNN-based graph propagation of feature values. This initialization and updating mechanism allows for informed feature learning and improved graph representation. We apply GraphPINE to cancer drug response prediction using drug screening and gene data collected for over 5,000 gene nodes included in a gene-gene graph with a drug-target interaction (DTI) graph for initial importance. The gene-gene graph and DTIs were obtained from curated sources and weighted by article count discussing relationships between drugs and genes. GraphPINE achieves a PR-AUC of 0.894 and ROC-AUC of 0.796 across 952 drugs. Code is available at https://anonymous.4open.science/r/GraphPINE-40DE.
drGAT: Attention-Guided Gene Assessment of Drug Response Utilizing a Drug-Cell-Gene Heterogeneous Network
May 14, 2024



Drug development is a lengthy process with a high failure rate. Increasingly, machine learning is utilized to facilitate the drug development processes. These models aim to enhance our understanding of drug characteristics, including their activity in biological contexts. However, a major challenge in drug response (DR) prediction is model interpretability as it aids in the validation of findings. This is important in biomedicine, where models need to be understandable in comparison with established knowledge of drug interactions with proteins. drGAT, a graph deep learning model, leverages a heterogeneous graph composed of relationships between proteins, cell lines, and drugs. drGAT is designed with two objectives: DR prediction as a binary sensitivity prediction and elucidation of drug mechanism from attention coefficients. drGAT has demonstrated superior performance over existing models, achieving 78\% accuracy (and precision), and 76\% F1 score for 269 DNA-damaging compounds of the NCI60 drug response dataset. To assess the model's interpretability, we conducted a review of drug-gene co-occurrences in Pubmed abstracts in comparison to the top 5 genes with the highest attention coefficients for each drug. We also examined whether known relationships were retained in the model by inspecting the neighborhoods of topoisomerase-related drugs. For example, our model retained TOP1 as a highly weighted predictive feature for irinotecan and topotecan, in addition to other genes that could potentially be regulators of the drugs. Our method can be used to accurately predict sensitivity to drugs and may be useful in the identification of biomarkers relating to the treatment of cancer patients.