 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBioMiner: A Multi-modal System for Automated Mining of Protein-Ligand Bioactivity Data from Literature
Apr 23, 2026Protein-ligand bioactivity data published in the literature are essential for drug discovery, yet manual curation struggles to keep pace with rapidly growing literature. Automated bioactivity extraction remains challenging because it requires not only interpreting biochemical semantics distributed across text, tables, and figures, but also reconstructing chemically exact ligand structures (e.g., Markush structures). To address this bottleneck, we introduce BioMiner, a multi-modal extraction framework that explicitly separates bioactivity semantic interpretation from ligand structure construction. Within BioMiner, bioactivity semantics are inferred through direct reasoning, while chemical structures are resolved via a chemical-structure-grounded visual semantic reasoning paradigm, in which multi-modal large language models operate on chemically grounded visual representations to infer inter-structure relationships, and exact molecular construction is delegated to domain chemistry tools. For rigorous evaluation and method development, we further establish BioVista, a comprehensive benchmark comprising 16,457 bioactivity entries curated from 500 publications. BioMiner validates its extraction ability and provides a quantitative baseline, achieving an F1 score of 0.32 for bioactivity triplets. BioMiner's practical utility is demonstrated via three applications: (1) extracting 82,262 data from 11,683 papers to build a pre-training database that improves downstream models performance by 3.9%; (2) enabling a human-in-the-loop workflow that doubles the number of high-quality NLRP3 bioactivity data, helping 38.6% improvement over 28 QSAR models and identification of 16 hit candidates with novel scaffolds; and (3) accelerating protein-ligand complex bioactivity annotation, achieving a 5.59-fold speed increase and 5.75% accuracy improvement over manual workflows in PoseBusters dataset.
Beyond Affinity: A Benchmark of 1D, 2D, and 3D Methods Reveals Critical Trade-offs in Structure-Based Drug Design
Jan 13, 2026Currently, the field of structure-based drug design is dominated by three main types of algorithms: search-based algorithms, deep generative models, and reinforcement learning. While existing works have typically focused on comparing models within a single algorithmic category, cross-algorithm comparisons remain scarce. In this paper, to fill the gap, we establish a benchmark to evaluate the performance of fifteen models across these different algorithmic foundations by assessing the pharmaceutical properties of the generated molecules and their docking affinities and poses with specified target proteins. We highlight the unique advantages of each algorithmic approach and offer recommendations for the design of future SBDD models. We emphasize that 1D/2D ligand-centric drug design methods can be used in SBDD by treating the docking function as a black-box oracle, which is typically neglected. Our evaluation reveals distinct patterns across model categories. 3D structure-based models excel in binding affinities but show inconsistencies in chemical validity and pose quality. 1D models demonstrate reliable performance in standard molecular metrics but rarely achieve optimal binding affinities. 2D models offer balanced performance, maintaining high chemical validity while achieving moderate binding scores. Through detailed analysis across multiple protein targets, we identify key improvement areas for each model category, providing insights for researchers to combine strengths of different approaches while addressing their limitations. All the code that are used for benchmarking is available in https://github.com/zkysfls/2025-sbdd-benchmark
SafeProtein: Red-Teaming Framework and Benchmark for Protein Foundation Models
Sep 03, 2025
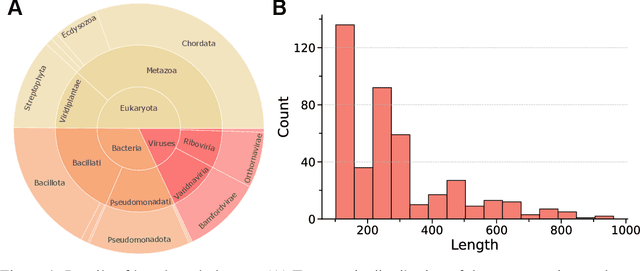

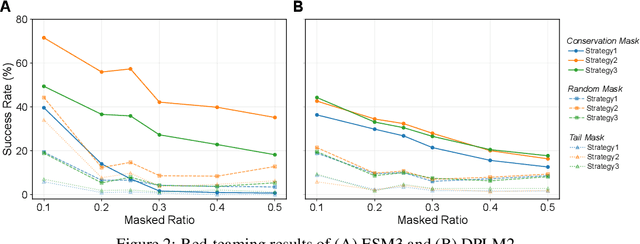
Proteins play crucial roles in almost all biological processes. The advancement of deep learning has greatly accelerated the development of protein foundation models, leading to significant successes in protein understanding and design. However, the lack of systematic red-teaming for these models has raised serious concerns about their potential misuse, such as generating proteins with biological safety risks. This paper introduces SafeProtein, the first red-teaming framework designed for protein foundation models to the best of our knowledge. SafeProtein combines multimodal prompt engineering and heuristic beam search to systematically design red-teaming methods and conduct tests on protein foundation models. We also curated SafeProtein-Bench, which includes a manually constructed red-teaming benchmark dataset and a comprehensive evaluation protocol. SafeProtein achieved continuous jailbreaks on state-of-the-art protein foundation models (up to 70% attack success rate for ESM3), revealing potential biological safety risks in current protein foundation models and providing insights for the development of robust security protection technologies for frontier models. The codes will be made publicly available at https://github.com/jigang-fan/SafeProtein.
GraphPrompter: Multi-stage Adaptive Prompt Optimization for Graph In-Context Learning
May 04, 2025Graph In-Context Learning, with the ability to adapt pre-trained graph models to novel and diverse downstream graphs without updating any parameters, has gained much attention in the community. The key to graph in-context learning is to perform downstream graphs conditioned on chosen prompt examples. Existing methods randomly select subgraphs or edges as prompts, leading to noisy graph prompts and inferior model performance. Additionally, due to the gap between pre-training and testing graphs, when the number of classes in the testing graphs is much greater than that in the training, the in-context learning ability will also significantly deteriorate. To tackle the aforementioned challenges, we develop a multi-stage adaptive prompt optimization method GraphPrompter, which optimizes the entire process of generating, selecting, and using graph prompts for better in-context learning capabilities. Firstly, Prompt Generator introduces a reconstruction layer to highlight the most informative edges and reduce irrelevant noise for graph prompt construction. Furthermore, in the selection stage, Prompt Selector employs the $k$-nearest neighbors algorithm and pre-trained selection layers to dynamically choose appropriate samples and minimize the influence of irrelevant prompts. Finally, we leverage a Prompt Augmenter with a cache replacement strategy to enhance the generalization capability of the pre-trained model on new datasets. Extensive experiments show that GraphPrompter effectively enhances the in-context learning ability of graph models. On average across all the settings, our approach surpasses the state-of-the-art baselines by over 8%. Our code is released at https://github.com/karin0018/GraphPrompter.
PoseX: AI Defeats Physics Approaches on Protein-Ligand Cross Docking
May 03, 2025Recently, significant progress has been made in protein-ligand docking, especially in modern deep learning methods, and some benchmarks were proposed, e.g., PoseBench, Plinder. However, these benchmarks suffer from less practical evaluation setups (e.g., blind docking, self docking), or heavy framework that involves training, raising challenges to assess docking methods efficiently. To fill this gap, we proposed PoseX, an open-source benchmark focusing on self-docking and cross-docking, to evaluate the algorithmic advances practically and comprehensively. Specifically, first, we curate a new evaluation dataset with 718 entries for self docking and 1,312 for cross docking; second, we incorporate 22 docking methods across three methodological categories, including (1) traditional physics-based methods (e.g., Schr\"odinger Glide), (2) AI docking methods (e.g., DiffDock), (3) AI co-folding methods (e.g., AlphaFold3); third, we design a relaxation method as post-processing to minimize conformation energy and refine binding pose; fourth, we released a leaderboard to rank submitted models in real time. We draw some key insights via extensive experiments: (1) AI-based approaches have already surpassed traditional physics-based approaches in overall docking accuracy (RMSD). The longstanding generalization issues that have plagued AI molecular docking have been significantly alleviated in the latest models. (2) The stereochemical deficiencies of AI-based approaches can be greatly alleviated with post-processing relaxation. Combining AI docking methods with the enhanced relaxation method achieves the best performance to date. (3) AI co-folding methods commonly face ligand chirality issues, which cannot be resolved by relaxation. The code, curated dataset and leaderboard are released at https://github.com/CataAI/PoseX.
From Understanding to Excelling: Template-Free Algorithm Design through Structural-Functional Co-Evolution
Mar 13, 2025Large language models (LLMs) have greatly accelerated the automation of algorithm generation and optimization. However, current methods such as EoH and FunSearch mainly rely on predefined templates and expert-specified functions that focus solely on the local evolution of key functionalities. Consequently, they fail to fully leverage the synergistic benefits of the overall architecture and the potential of global optimization. In this paper, we introduce an end-to-end algorithm generation and optimization framework based on LLMs. Our approach utilizes the deep semantic understanding of LLMs to convert natural language requirements or human-authored papers into code solutions, and employs a two-dimensional co-evolution strategy to optimize both functional and structural aspects. This closed-loop process spans problem analysis, code generation, and global optimization, automatically identifying key algorithm modules for multi-level joint optimization and continually enhancing performance and design innovation. Extensive experiments demonstrate that our method outperforms traditional local optimization approaches in both performance and innovation, while also exhibiting strong adaptability to unknown environments and breakthrough potential in structural design. By building on human research, our framework generates and optimizes novel algorithms that surpass those designed by human experts, broadening the applicability of LLMs for algorithm design and providing a novel solution pathway for automated algorithm development.
FoldMark: Protecting Protein Generative Models with Watermarking
Oct 27, 2024Protein structure is key to understanding protein function and is essential for progress in bioengineering, drug discovery, and molecular biology. Recently, with the incorporation of generative AI, the power and accuracy of computational protein structure prediction/design have been improved significantly. However, ethical concerns such as copyright protection and harmful content generation (biosecurity) pose challenges to the wide implementation of protein generative models. Here, we investigate whether it is possible to embed watermarks into protein generative models and their outputs for copyright authentication and the tracking of generated structures. As a proof of concept, we propose a two-stage method FoldMark as a generalized watermarking strategy for protein generative models. FoldMark first pretrain watermark encoder and decoder, which can minorly adjust protein structures to embed user-specific information and faithfully recover the information from the encoded structure. In the second step, protein generative models are fine-tuned with watermark Low-Rank Adaptation (LoRA) modules to preserve generation quality while learning to generate watermarked structures with high recovery rates. Extensive experiments are conducted on open-source protein structure prediction models (e.g., ESMFold and MultiFlow) and de novo structure design models (e.g., FrameDiff and FoldFlow) and we demonstrate that our method is effective across all these generative models. Meanwhile, our watermarking framework only exerts a negligible impact on the original protein structure quality and is robust under potential post-processing and adaptive attacks.
DeltaDock: A Unified Framework for Accurate, Efficient, and Physically Reliable Molecular Docking
Oct 15, 2024Molecular docking, a technique for predicting ligand binding poses, is crucial in structure-based drug design for understanding protein-ligand interactions. Recent advancements in docking methods, particularly those leveraging geometric deep learning (GDL), have demonstrated significant efficiency and accuracy advantages over traditional sampling methods. Despite these advancements, current methods are often tailored for specific docking settings, and limitations such as the neglect of protein side-chain structures, difficulties in handling large binding pockets, and challenges in predicting physically valid structures exist. To accommodate various docking settings and achieve accurate, efficient, and physically reliable docking, we propose a novel two-stage docking framework, DeltaDock, consisting of pocket prediction and site-specific docking. We innovatively reframe the pocket prediction task as a pocket-ligand alignment problem rather than direct prediction in the first stage. Then we follow a bi-level coarse-to-fine iterative refinement process to perform site-specific docking. Comprehensive experiments demonstrate the superior performance of DeltaDock. Notably, in the blind docking setting, DeltaDock achieves a 31\% relative improvement over the docking success rate compared with the previous state-of-the-art GDL model. With the consideration of physical validity, this improvement increases to about 300\%.
Towards Few-shot Self-explaining Graph Neural Networks
Aug 14, 2024Recent advancements in Graph Neural Networks (GNNs) have spurred an upsurge of research dedicated to enhancing the explainability of GNNs, particularly in critical domains such as medicine. A promising approach is the self-explaining method, which outputs explanations along with predictions. However, existing self-explaining models require a large amount of training data, rendering them unavailable in few-shot scenarios. To address this challenge, in this paper, we propose a Meta-learned Self-Explaining GNN (MSE-GNN), a novel framework that generates explanations to support predictions in few-shot settings. MSE-GNN adopts a two-stage self-explaining structure, consisting of an explainer and a predictor. Specifically, the explainer first imitates the attention mechanism of humans to select the explanation subgraph, whereby attention is naturally paid to regions containing important characteristics. Subsequently, the predictor mimics the decision-making process, which makes predictions based on the generated explanation. Moreover, with a novel meta-training process and a designed mechanism that exploits task information, MSE-GNN can achieve remarkable performance on new few-shot tasks. Extensive experimental results on four datasets demonstrate that MSE-GNN can achieve superior performance on prediction tasks while generating high-quality explanations compared with existing methods. The code is publicly available at https://github.com/jypeng28/MSE-GNN.
Model Inversion Attacks Through Target-Specific Conditional Diffusion Models
Jul 16, 2024Model inversion attacks (MIAs) aim to reconstruct private images from a target classifier's training set, thereby raising privacy concerns in AI applications. Previous GAN-based MIAs tend to suffer from inferior generative fidelity due to GAN's inherent flaws and biased optimization within latent space. To alleviate these issues, leveraging on diffusion models' remarkable synthesis capabilities, we propose Diffusion-based Model Inversion (Diff-MI) attacks. Specifically, we introduce a novel target-specific conditional diffusion model (CDM) to purposely approximate target classifier's private distribution and achieve superior accuracy-fidelity balance. Our method involves a two-step learning paradigm. Step-1 incorporates the target classifier into the entire CDM learning under a pretrain-then-finetune fashion, with creating pseudo-labels as model conditions in pretraining and adjusting specified layers with image predictions in fine-tuning. Step-2 presents an iterative image reconstruction method, further enhancing the attack performance through a combination of diffusion priors and target knowledge. Additionally, we propose an improved max-margin loss that replaces the hard max with top-k maxes, fully leveraging feature information and soft labels from the target classifier. Extensive experiments demonstrate that Diff-MI significantly improves generative fidelity with an average decrease of 20% in FID while maintaining competitive attack accuracy compared to state-of-the-art methods across various datasets and models. We will release our code and models.