 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecular Representations in Implicit Functional Space via Hyper-Networks
Jan 29, 2026Molecular representations fundamentally shape how machine learning systems reason about molecular structure and physical properties. Most existing approaches adopt a discrete pipeline: molecules are encoded as sequences, graphs, or point clouds, mapped to fixed-dimensional embeddings, and then used for task-specific prediction. This paradigm treats molecules as discrete objects, despite their intrinsically continuous and field-like physical nature. We argue that molecular learning can instead be formulated as learning in function space. Specifically, we model each molecule as a continuous function over three-dimensional (3D) space and treat this molecular field as the primary object of representation. From this perspective, conventional molecular representations arise as particular sampling schemes of an underlying continuous object. We instantiate this formulation with MolField, a hyper-network-based framework that learns distributions over molecular fields. To ensure physical consistency, these functions are defined over canonicalized coordinates, yielding invariance to global SE(3) transformations. To enable learning directly over functions, we introduce a structured weight tokenization and train a sequence-based hyper-network to model a shared prior over molecular fields. We evaluate MolField on molecular dynamics and property prediction. Our results show that treating molecules as continuous functions fundamentally changes how molecular representations generalize across tasks and yields downstream behavior that is stable to how molecules are discretized or queried.
SafeProtein: Red-Teaming Framework and Benchmark for Protein Foundation Models
Sep 03, 2025
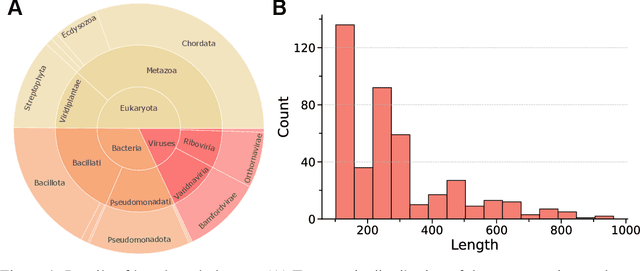

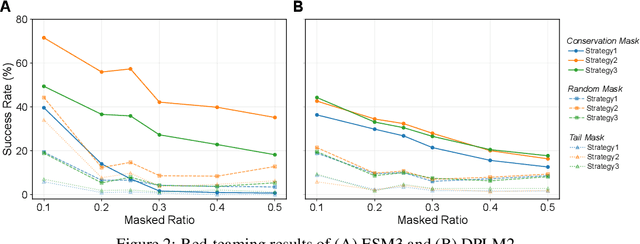
Proteins play crucial roles in almost all biological processes. The advancement of deep learning has greatly accelerated the development of protein foundation models, leading to significant successes in protein understanding and design. However, the lack of systematic red-teaming for these models has raised serious concerns about their potential misuse, such as generating proteins with biological safety risks. This paper introduces SafeProtein, the first red-teaming framework designed for protein foundation models to the best of our knowledge. SafeProtein combines multimodal prompt engineering and heuristic beam search to systematically design red-teaming methods and conduct tests on protein foundation models. We also curated SafeProtein-Bench, which includes a manually constructed red-teaming benchmark dataset and a comprehensive evaluation protocol. SafeProtein achieved continuous jailbreaks on state-of-the-art protein foundation models (up to 70% attack success rate for ESM3), revealing potential biological safety risks in current protein foundation models and providing insights for the development of robust security protection technologies for frontier models. The codes will be made publicly available at https://github.com/jigang-fan/SafeProtein.
Genome-Bench: A Scientific Reasoning Benchmark from Real-World Expert Discussions
May 26, 2025In this short report, we present an automated pipeline tailored for the genomics domain and introduce \textit{Genome-Bench}, a new benchmark constructed from over a decade of scientific forum discussions on genome engineering. Our pipeline transforms raw interactions into a reinforcement learning friendly multiple-choice questions format, supported by 3000+ high quality question answer pairs spanning foundational biology, experimental troubleshooting, tool usage, and beyond. To our knowledge, this is the first end-to-end pipeline for teaching LLMs to reason from scientific discussions, with promising potential for generalization across scientific domains beyond biology.
FoldMark: Protecting Protein Generative Models with Watermarking
Oct 27, 2024Protein structure is key to understanding protein function and is essential for progress in bioengineering, drug discovery, and molecular biology. Recently, with the incorporation of generative AI, the power and accuracy of computational protein structure prediction/design have been improved significantly. However, ethical concerns such as copyright protection and harmful content generation (biosecurity) pose challenges to the wide implementation of protein generative models. Here, we investigate whether it is possible to embed watermarks into protein generative models and their outputs for copyright authentication and the tracking of generated structures. As a proof of concept, we propose a two-stage method FoldMark as a generalized watermarking strategy for protein generative models. FoldMark first pretrain watermark encoder and decoder, which can minorly adjust protein structures to embed user-specific information and faithfully recover the information from the encoded structure. In the second step, protein generative models are fine-tuned with watermark Low-Rank Adaptation (LoRA) modules to preserve generation quality while learning to generate watermarked structures with high recovery rates. Extensive experiments are conducted on open-source protein structure prediction models (e.g., ESMFold and MultiFlow) and de novo structure design models (e.g., FrameDiff and FoldFlow) and we demonstrate that our method is effective across all these generative models. Meanwhile, our watermarking framework only exerts a negligible impact on the original protein structure quality and is robust under potential post-processing and adaptive attacks.
Latent Diffusion Models for Controllable RNA Sequence Generation
Sep 15, 2024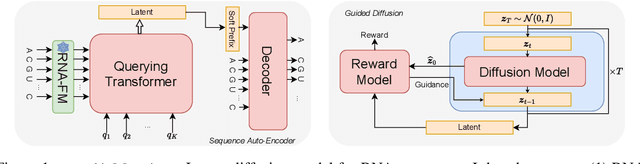
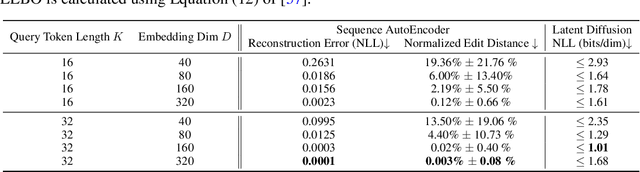

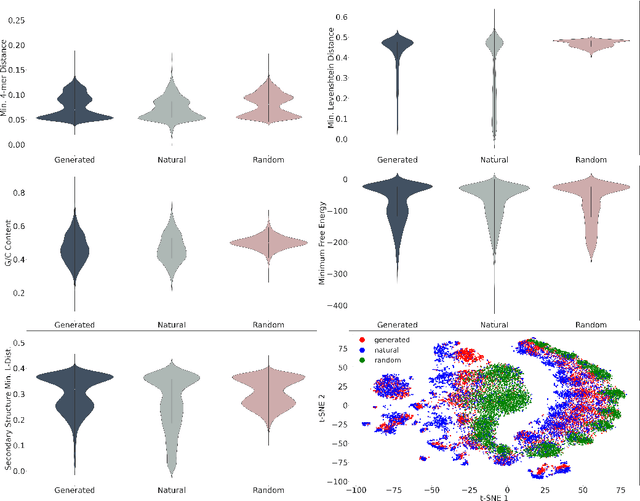
This paper presents RNAdiffusion, a latent diffusion model for generating and optimizing discrete RNA sequences. RNA is a particularly dynamic and versatile molecule in biological processes. RNA sequences exhibit high variability and diversity, characterized by their variable lengths, flexible three-dimensional structures, and diverse functions. We utilize pretrained BERT-type models to encode raw RNAs into token-level biologically meaningful representations. A Q-Former is employed to compress these representations into a fixed-length set of latent vectors, with an autoregressive decoder trained to reconstruct RNA sequences from these latent variables. We then develop a continuous diffusion model within this latent space. To enable optimization, we train reward networks to estimate functional properties of RNA from the latent variables. We employ gradient-based guidance during the backward diffusion process, aiming to generate RNA sequences that are optimized for higher rewards. Empirical experiments confirm that RNAdiffusion generates non-coding RNAs that align with natural distributions across various biological indicators. We fine-tuned the diffusion model on untranslated regions (UTRs) of mRNA and optimize sample sequences for protein translation efficiencies. Our guided diffusion model effectively generates diverse UTR sequences with high Mean Ribosome Loading (MRL) and Translation Efficiency (TE), surpassing baselines. These results hold promise for studies on RNA sequence-function relationships, protein synthesis, and enhancing therapeutic RNA design.
CRISPR-GPT: An LLM Agent for Automated Design of Gene-Editing Experiments
Apr 27, 2024The introduction of genome engineering technology has transformed biomedical research, making it possible to make precise changes to genetic information. However, creating an efficient gene-editing system requires a deep understanding of CRISPR technology, and the complex experimental systems under investigation. While Large Language Models (LLMs) have shown promise in various tasks, they often lack specific knowledge and struggle to accurately solve biological design problems. In this work, we introduce CRISPR-GPT, an LLM agent augmented with domain knowledge and external tools to automate and enhance the design process of CRISPR-based gene-editing experiments. CRISPR-GPT leverages the reasoning ability of LLMs to facilitate the process of selecting CRISPR systems, designing guide RNAs, recommending cellular delivery methods, drafting protocols, and designing validation experiments to confirm editing outcomes. We showcase the potential of CRISPR-GPT for assisting non-expert researchers with gene-editing experiments from scratch and validate the agent's effectiveness in a real-world use case. Furthermore, we explore the ethical and regulatory considerations associated with automated gene-editing design, highlighting the need for responsible and transparent use of these tools. Our work aims to bridge the gap between beginner biological researchers and CRISPR genome engineering techniques, and demonstrate the potential of LLM agents in facilitating complex biological discovery tasks.
A 5' UTR Language Model for Decoding Untranslated Regions of mRNA and Function Predictions
Oct 06, 2023The 5' UTR, a regulatory region at the beginning of an mRNA molecule, plays a crucial role in regulating the translation process and impacts the protein expression level. Language models have showcased their effectiveness in decoding the functions of protein and genome sequences. Here, we introduced a language model for 5' UTR, which we refer to as the UTR-LM. The UTR-LM is pre-trained on endogenous 5' UTRs from multiple species and is further augmented with supervised information including secondary structure and minimum free energy. We fine-tuned the UTR-LM in a variety of downstream tasks. The model outperformed the best-known benchmark by up to 42% for predicting the Mean Ribosome Loading, and by up to 60% for predicting the Translation Efficiency and the mRNA Expression Level. The model also applies to identifying unannotated Internal Ribosome Entry Sites within the untranslated region and improves the AUPR from 0.37 to 0.52 compared to the best baseline. Further, we designed a library of 211 novel 5' UTRs with high predicted values of translation efficiency and evaluated them via a wet-lab assay. Experiment results confirmed that our top designs achieved a 32.5% increase in protein production level relative to well-established 5' UTR optimized for therapeutics.
Bandit Theory and Thompson Sampling-Guided Directed Evolution for Sequence Optimization
Jun 05, 2022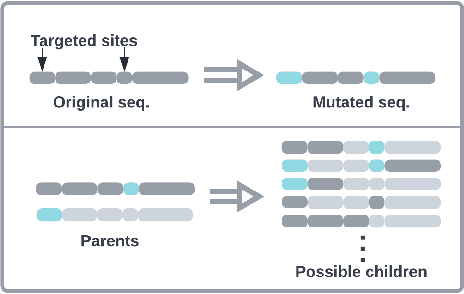
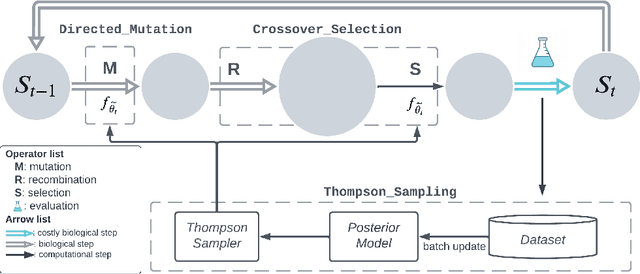
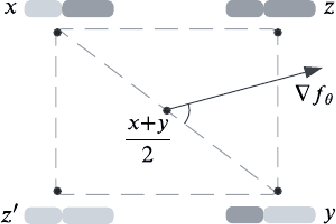
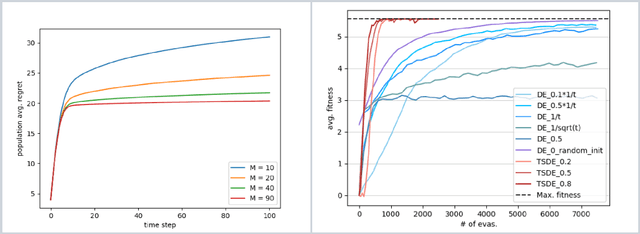
Directed Evolution (DE), a landmark wet-lab method originated in 1960s, enables discovery of novel protein designs via evolving a population of candidate sequences. Recent advances in biotechnology has made it possible to collect high-throughput data, allowing the use of machine learning to map out a protein's sequence-to-function relation. There is a growing interest in machine learning-assisted DE for accelerating protein optimization. Yet the theoretical understanding of DE, as well as the use of machine learning in DE, remains limited. In this paper, we connect DE with the bandit learning theory and make a first attempt to study regret minimization in DE. We propose a Thompson Sampling-guided Directed Evolution (TS-DE) framework for sequence optimization, where the sequence-to-function mapping is unknown and querying a single value is subject to costly and noisy measurements. TS-DE updates a posterior of the function based on collected measurements. It uses a posterior-sampled function estimate to guide the crossover recombination and mutation steps in DE. In the case of a linear model, we show that TS-DE enjoys a Bayesian regret of order $\tilde O(d^{2}\sqrt{MT})$, where $d$ is feature dimension, $M$ is population size and $T$ is number of rounds. This regret bound is nearly optimal, confirming that bandit learning can provably accelerate DE. It may have implications for more general sequence optimization and evolutionary algorithms.