 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolAct: An Agentic RL Framework for Molecular Editing and Property Optimization
Dec 24, 2025Molecular editing and optimization are multi-step problems that require iteratively improving properties while keeping molecules chemically valid and structurally similar. We frame both tasks as sequential, tool-guided decisions and introduce MolAct, an agentic reinforcement learning framework that employs a two-stage training paradigm: first building editing capability, then optimizing properties while reusing the learned editing behaviors. To the best of our knowledge, this is the first work to formalize molecular design as an Agentic Reinforcement Learning problem, where an LLM agent learns to interleave reasoning, tool-use, and molecular optimization. The framework enables agents to interact in multiple turns, invoking chemical tools for validity checking, property assessment, and similarity control, and leverages their feedback to refine subsequent edits. We instantiate the MolAct framework to train two model families: MolEditAgent for molecular editing tasks and MolOptAgent for molecular optimization tasks. In molecular editing, MolEditAgent-7B delivers 100, 95, and 98 valid add, delete, and substitute edits, outperforming strong closed "thinking" baselines such as DeepSeek-R1; MolEditAgent-3B approaches the performance of much larger open "thinking" models like Qwen3-32B-think. In molecular optimization, MolOptAgent-7B (trained on MolEditAgent-7B) surpasses the best closed "thinking" baseline (e.g., Claude 3.7) on LogP and remains competitive on solubility, while maintaining balanced performance across other objectives. These results highlight that treating molecular design as a multi-step, tool-augmented process is key to reliable and interpretable improvements.
An Agentic Framework for Autonomous Materials Computation
Dec 22, 2025



Large Language Models (LLMs) have emerged as powerful tools for accelerating scientific discovery, yet their static knowledge and hallucination issues hinder autonomous research applications. Recent advances integrate LLMs into agentic frameworks, enabling retrieval, reasoning, and tool use for complex scientific workflows. Here, we present a domain-specialized agent designed for reliable automation of first-principles materials computations. By embedding domain expertise, the agent ensures physically coherent multi-step workflows and consistently selects convergent, well-posed parameters, thereby enabling reliable end-to-end computational execution. A new benchmark of diverse computational tasks demonstrates that our system significantly outperforms standalone LLMs in both accuracy and robustness. This work establishes a verifiable foundation for autonomous computational experimentation and represents a key step toward fully automated scientific discovery.
Revisiting the Broken Symmetry Phase of Solid Hydrogen: A Neural Network Variational Monte Carlo Study
Dec 19, 2025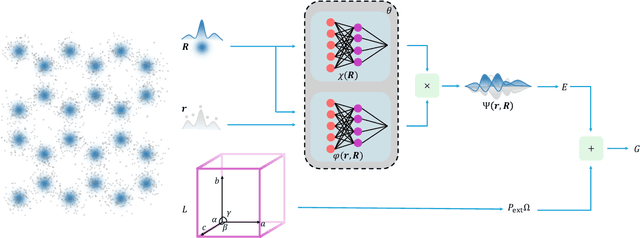

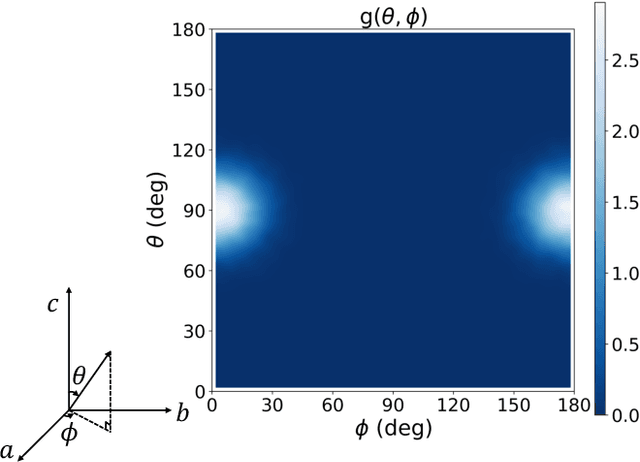
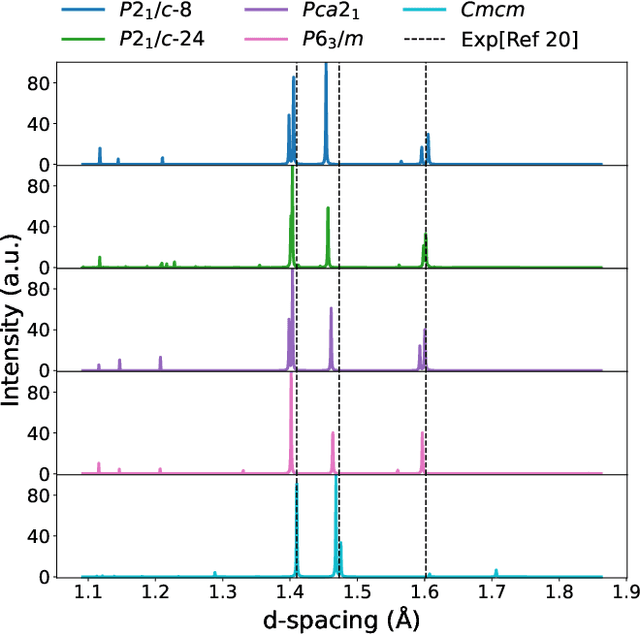
The crystal structure of high-pressure solid hydrogen remains a fundamental open problem. Although the research frontier has mostly shifted toward ultra-high pressure phases above 400 GPa, we show that even the broken symmetry phase observed around 130~GPa requires revisiting due to its intricate coupling of electronic and nuclear degrees of freedom. Here, we develop a first principle quantum Monte Carlo framework based on a deep neural network wave function that treats both electrons and nuclei quantum mechanically within the constant pressure ensemble. Our calculations reveal an unreported ground-state structure candidate for the broken symmetry phase with $Cmcm$ space group symmetry, and we test its stability up to 96 atoms. The predicted structure quantitatively matches the experimental equation of state and X-ray diffraction patterns. Furthermore, our group-theoretical analysis shows that the $Cmcm$ structure is compatible with existing Raman and infrared spectroscopic data. Crucially, static density functional theory calculation reveals the $Cmcm$ structure as a dynamically unstable saddle point on the Born-Oppenheimer potential energy surface, demonstrating that a full quantum many-body treatment of the problem is necessary. These results shed new light on the phase diagram of high-pressure hydrogen and call for further experimental verifications.
Probing Scientific General Intelligence of LLMs with Scientist-Aligned Workflows
Dec 18, 2025Despite advances in scientific AI, a coherent framework for Scientific General Intelligence (SGI)-the ability to autonomously conceive, investigate, and reason across scientific domains-remains lacking. We present an operational SGI definition grounded in the Practical Inquiry Model (PIM: Deliberation, Conception, Action, Perception) and operationalize it via four scientist-aligned tasks: deep research, idea generation, dry/wet experiments, and experimental reasoning. SGI-Bench comprises over 1,000 expert-curated, cross-disciplinary samples inspired by Science's 125 Big Questions, enabling systematic evaluation of state-of-the-art LLMs. Results reveal gaps: low exact match (10--20%) in deep research despite step-level alignment; ideas lacking feasibility and detail; high code executability but low execution result accuracy in dry experiments; low sequence fidelity in wet protocols; and persistent multimodal comparative-reasoning challenges. We further introduce Test-Time Reinforcement Learning (TTRL), which optimizes retrieval-augmented novelty rewards at inference, enhancing hypothesis novelty without reference answer. Together, our PIM-grounded definition, workflow-centric benchmark, and empirical insights establish a foundation for AI systems that genuinely participate in scientific discovery.
ATLAS: A High-Difficulty, Multidisciplinary Benchmark for Frontier Scientific Reasoning
Nov 18, 2025The rapid advancement of Large Language Models (LLMs) has led to performance saturation on many established benchmarks, questioning their ability to distinguish frontier models. Concurrently, existing high-difficulty benchmarks often suffer from narrow disciplinary focus, oversimplified answer formats, and vulnerability to data contamination, creating a fidelity gap with real-world scientific inquiry. To address these challenges, we introduce ATLAS (AGI-Oriented Testbed for Logical Application in Science), a large-scale, high-difficulty, and cross-disciplinary evaluation suite composed of approximately 800 original problems. Developed by domain experts (PhD-level and above), ATLAS spans seven core scientific fields: mathematics, physics, chemistry, biology, computer science, earth science, and materials science. Its key features include: (1) High Originality and Contamination Resistance, with all questions newly created or substantially adapted to prevent test data leakage; (2) Cross-Disciplinary Focus, designed to assess models' ability to integrate knowledge and reason across scientific domains; (3) High-Fidelity Answers, prioritizing complex, open-ended answers involving multi-step reasoning and LaTeX-formatted expressions over simple multiple-choice questions; and (4) Rigorous Quality Control, employing a multi-stage process of expert peer review and adversarial testing to ensure question difficulty, scientific value, and correctness. We also propose a robust evaluation paradigm using a panel of LLM judges for automated, nuanced assessment of complex answers. Preliminary results on leading models demonstrate ATLAS's effectiveness in differentiating their advanced scientific reasoning capabilities. We plan to develop ATLAS into a long-term, open, community-driven platform to provide a reliable "ruler" for progress toward Artificial General Intelligence.
InertialAR: Autoregressive 3D Molecule Generation with Inertial Frames
Oct 31, 2025Transformer-based autoregressive models have emerged as a unifying paradigm across modalities such as text and images, but their extension to 3D molecule generation remains underexplored. The gap stems from two fundamental challenges: (1) tokenizing molecules into a canonical 1D sequence of tokens that is invariant to both SE(3) transformations and atom index permutations, and (2) designing an architecture capable of modeling hybrid atom-based tokens that couple discrete atom types with continuous 3D coordinates. To address these challenges, we introduce InertialAR. InertialAR devises a canonical tokenization that aligns molecules to their inertial frames and reorders atoms to ensure SE(3) and permutation invariance. Moreover, InertialAR equips the attention mechanism with geometric awareness via geometric rotary positional encoding (GeoRoPE). In addition, it utilizes a hierarchical autoregressive paradigm to predict the next atom-based token, predicting the atom type first and then its 3D coordinates via Diffusion loss. Experimentally, InertialAR achieves state-of-the-art performance on 7 of the 10 evaluation metrics for unconditional molecule generation across QM9, GEOM-Drugs, and B3LYP. Moreover, it significantly outperforms strong baselines in controllable generation for targeted chemical functionality, attaining state-of-the-art results across all 5 metrics.
ChemBOMAS: Accelerated BO in Chemistry with LLM-Enhanced Multi-Agent System
Sep 10, 2025The efficiency of Bayesian optimization (BO) in chemistry is often hindered by sparse experimental data and complex reaction mechanisms. To overcome these limitations, we introduce ChemBOMAS, a new framework named LLM-Enhanced Multi-Agent System for accelerating BO in chemistry. ChemBOMAS's optimization process is enhanced by LLMs and synergistically employs two strategies: knowledge-driven coarse-grained optimization and data-driven fine-grained optimization. First, in the knowledge-driven coarse-grained optimization stage, LLMs intelligently decompose the vast search space by reasoning over existing chemical knowledge to identify promising candidate regions. Subsequently, in the data-driven fine-grained optimization stage, LLMs enhance the BO process within these candidate regions by generating pseudo-data points, thereby improving data utilization efficiency and accelerating convergence. Benchmark evaluations** further confirm that ChemBOMAS significantly enhances optimization effectiveness and efficiency compared to various BO algorithms. Importantly, the practical utility of ChemBOMAS was validated through wet-lab experiments conducted under pharmaceutical industry protocols, targeting conditional optimization for a previously unreported and challenging chemical reaction. In the wet experiment, ChemBOMAS achieved an optimal objective value of 96%. This was substantially higher than the 15% achieved by domain experts. This real-world success, together with strong performance on benchmark evaluations, highlights ChemBOMAS as a powerful tool to accelerate chemical discovery.
A Survey of Scientific Large Language Models: From Data Foundations to Agent Frontiers
Aug 28, 2025



Scientific Large Language Models (Sci-LLMs) are transforming how knowledge is represented, integrated, and applied in scientific research, yet their progress is shaped by the complex nature of scientific data. This survey presents a comprehensive, data-centric synthesis that reframes the development of Sci-LLMs as a co-evolution between models and their underlying data substrate. We formulate a unified taxonomy of scientific data and a hierarchical model of scientific knowledge, emphasizing the multimodal, cross-scale, and domain-specific challenges that differentiate scientific corpora from general natural language processing datasets. We systematically review recent Sci-LLMs, from general-purpose foundations to specialized models across diverse scientific disciplines, alongside an extensive analysis of over 270 pre-/post-training datasets, showing why Sci-LLMs pose distinct demands -- heterogeneous, multi-scale, uncertainty-laden corpora that require representations preserving domain invariance and enabling cross-modal reasoning. On evaluation, we examine over 190 benchmark datasets and trace a shift from static exams toward process- and discovery-oriented assessments with advanced evaluation protocols. These data-centric analyses highlight persistent issues in scientific data development and discuss emerging solutions involving semi-automated annotation pipelines and expert validation. Finally, we outline a paradigm shift toward closed-loop systems where autonomous agents based on Sci-LLMs actively experiment, validate, and contribute to a living, evolving knowledge base. Collectively, this work provides a roadmap for building trustworthy, continually evolving artificial intelligence (AI) systems that function as a true partner in accelerating scientific discovery.
CMPhysBench: A Benchmark for Evaluating Large Language Models in Condensed Matter Physics
Aug 25, 2025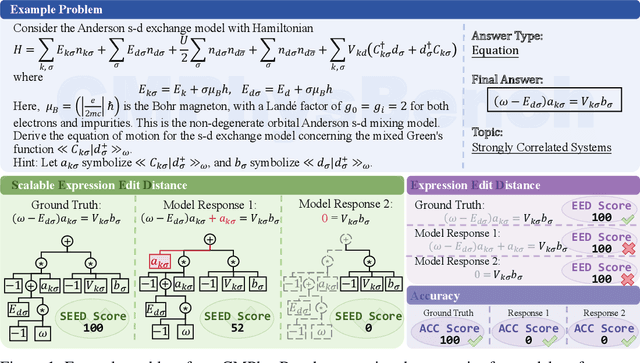
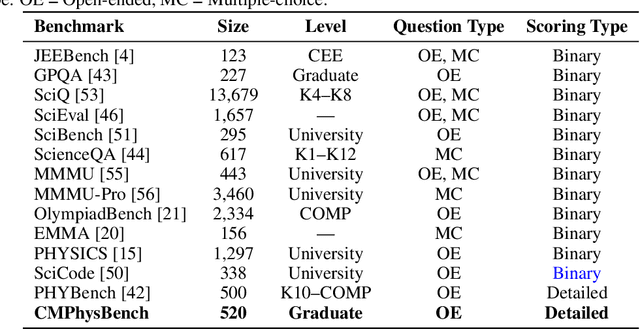
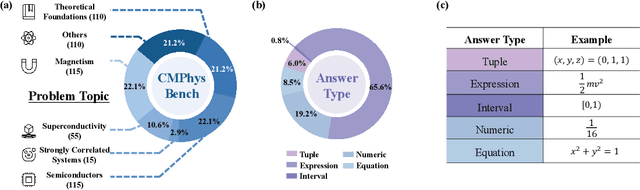
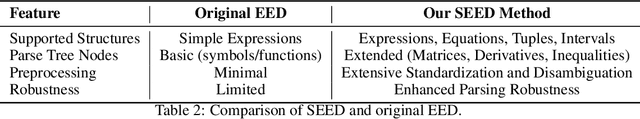
We introduce CMPhysBench, designed to assess the proficiency of Large Language Models (LLMs) in Condensed Matter Physics, as a novel Benchmark. CMPhysBench is composed of more than 520 graduate-level meticulously curated questions covering both representative subfields and foundational theoretical frameworks of condensed matter physics, such as magnetism, superconductivity, strongly correlated systems, etc. To ensure a deep understanding of the problem-solving process,we focus exclusively on calculation problems, requiring LLMs to independently generate comprehensive solutions. Meanwhile, leveraging tree-based representations of expressions, we introduce the Scalable Expression Edit Distance (SEED) score, which provides fine-grained (non-binary) partial credit and yields a more accurate assessment of similarity between prediction and ground-truth. Our results show that even the best models, Grok-4, reach only 36 average SEED score and 28% accuracy on CMPhysBench, underscoring a significant capability gap, especially for this practical and frontier domain relative to traditional physics. The code anddataset are publicly available at https://github.com/CMPhysBench/CMPhysBench.
Iterative Pretraining Framework for Interatomic Potentials
Jul 27, 2025Machine learning interatomic potentials (MLIPs) enable efficient molecular dynamics (MD) simulations with ab initio accuracy and have been applied across various domains in physical science. However, their performance often relies on large-scale labeled training data. While existing pretraining strategies can improve model performance, they often suffer from a mismatch between the objectives of pretraining and downstream tasks or rely on extensive labeled datasets and increasingly complex architectures to achieve broad generalization. To address these challenges, we propose Iterative Pretraining for Interatomic Potentials (IPIP), a framework designed to iteratively improve the predictive performance of MLIP models. IPIP incorporates a forgetting mechanism to prevent iterative training from converging to suboptimal local minima. Unlike general-purpose foundation models, which frequently underperform on specialized tasks due to a trade-off between generality and system-specific accuracy, IPIP achieves higher accuracy and efficiency using lightweight architectures. Compared to general-purpose force fields, this approach achieves over 80% reduction in prediction error and up to 4x speedup in the challenging Mo-S-O system, enabling fast and accurate simulations.