 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGenerative structural elucidation from mass spectra as an iterative optimization problem
Feb 07, 2026Liquid chromatography tandem mass spectrometry (LC-MS/MS) is a critical analytical technique for molecular identification across metabolomics, environmental chemistry, and chemical forensics. A variety of computational methods have emerged for structural annotation of spectral features of interest, but many of these features cannot be confidently annotated with reference structures or spectra. Here, we introduce FOAM (Formula-constrained Optimization for Annotating Metabolites), a computational workflow that poses structure elucidation from LC-MS/MS as an iterative optimization problem. FOAM couples a formula-constrained graph genetic algorithm with spectral simulation to explore candidate annotations given an experimental spectrum. We demonstrate FOAM's performance on the NIST'20 and MassSpecGym datasets as both a standalone elucidation pipeline and as a complement to existing inverse models. This work establishes iterative optimization as an effective and extensible paradigm for structural elucidation.
Neural Graph Matching Improves Retrieval Augmented Generation in Molecular Machine Learning
Feb 25, 2025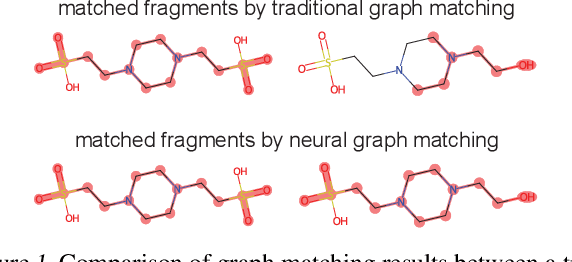
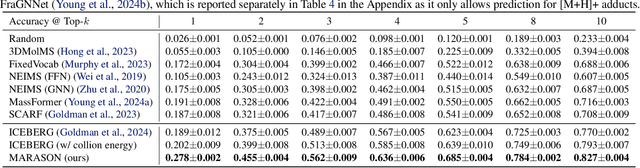
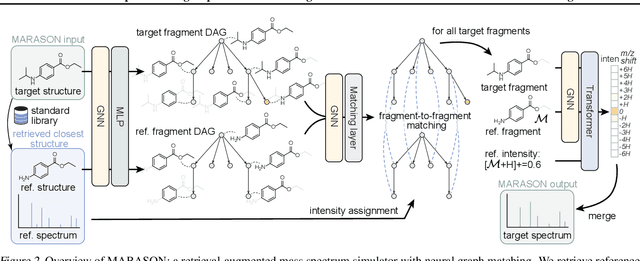
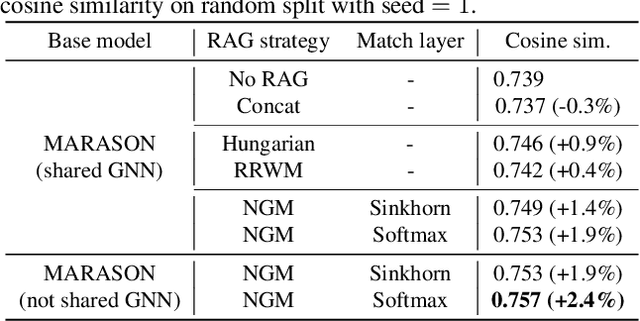
Molecular machine learning has gained popularity with the advancements of geometric deep learning. In parallel, retrieval-augmented generation has become a principled approach commonly used with language models. However, the optimal integration of retrieval augmentation into molecular machine learning remains unclear. Graph neural networks stand to benefit from clever matching to understand the structural alignment of retrieved molecules to a query molecule. Neural graph matching offers a compelling solution by explicitly modeling node and edge affinities between two structural graphs while employing a noise-robust, end-to-end neural network to learn affinity metrics. We apply this approach to mass spectrum simulation and introduce MARASON, a novel model that incorporates neural graph matching to enhance a fragmentation-based neural network. Experimental results highlight the effectiveness of our design, with MARASON achieving 28% top-1 accuracy, a substantial improvement over the non-retrieval state-of-the-art accuracy of 19%. Moreover, MARASON outperforms both naive retrieval-augmented generation methods and traditional graph matching approaches.
Electron flow matching for generative reaction mechanism prediction obeying conservation laws
Feb 18, 2025Central to our understanding of chemical reactivity is the principle of mass conservation, which is fundamental for ensuring physical consistency, balancing equations, and guiding reaction design. However, data-driven computational models for tasks such as reaction product prediction rarely abide by this most basic constraint. In this work, we recast the problem of reaction prediction as a problem of electron redistribution using the modern deep generative framework of flow matching. Our model, FlowER, overcomes limitations inherent in previous approaches by enforcing exact mass conservation, thereby resolving hallucinatory failure modes, recovering mechanistic reaction sequences for unseen substrate scaffolds, and generalizing effectively to out-of-domain reaction classes with extremely data-efficient fine-tuning. FlowER additionally enables estimation of thermodynamic or kinetic feasibility and manifests a degree of chemical intuition in reaction prediction tasks. This inherently interpretable framework represents a significant step in bridging the gap between predictive accuracy and mechanistic understanding in data-driven reaction outcome prediction.
The impact of conformer quality on learned representations of molecular conformer ensembles
Feb 18, 2025Training machine learning models to predict properties of molecular conformer ensembles is an increasingly popular strategy to accelerate the conformational analysis of drug-like small molecules, reactive organic substrates, and homogeneous catalysts. For high-throughput analyses especially, trained surrogate models can help circumvent traditional approaches to conformational analysis that rely on expensive conformer searches and geometry optimizations. Here, we question how the performance of surrogate models for predicting 3D conformer-dependent properties (of a single, active conformer) is affected by the quality of the 3D conformers used as their input. How well do lower-quality conformers inform the prediction of properties of higher-quality conformers? Does the fidelity of geometry optimization matter when encoding random conformers? For models that encode sets of conformers, how does the presence of the active conformer that induces the target property affect model accuracy? How do predictions from a surrogate model compare to estimating the properties from cheap ensembles themselves? We explore these questions in the context of predicting Sterimol parameters of conformer ensembles optimized with density functional theory. Although answers will be case-specific, our analyses provide a valuable perspective on 3D representation learning models and raise practical considerations regarding when conformer quality matters.
DiffMS: Diffusion Generation of Molecules Conditioned on Mass Spectra
Feb 13, 2025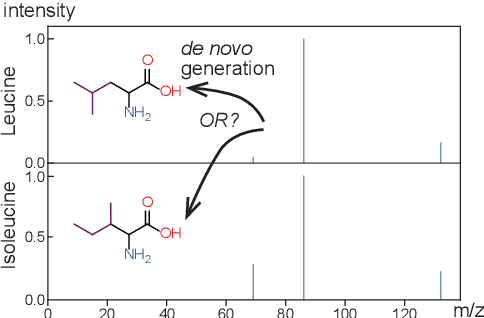
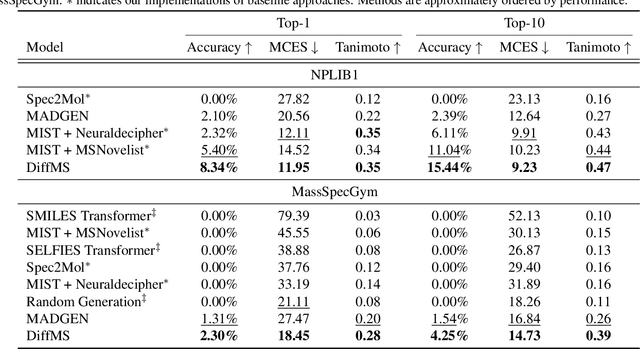
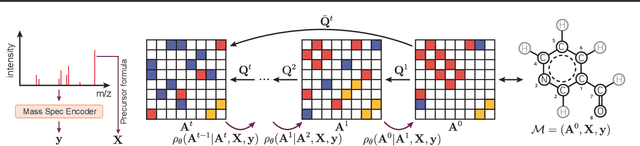
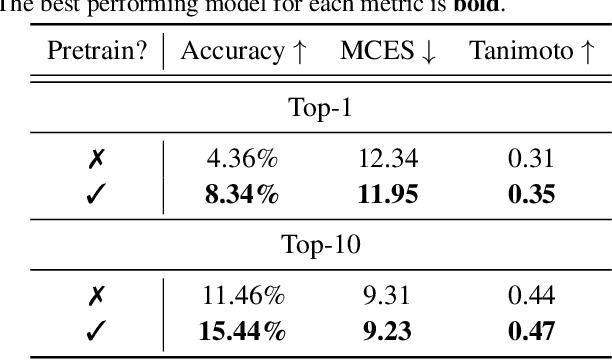
Mass spectrometry plays a fundamental role in elucidating the structures of unknown molecules and subsequent scientific discoveries. One formulation of the structure elucidation task is the conditional $\textit{de novo}$ generation of molecular structure given a mass spectrum. Toward a more accurate and efficient scientific discovery pipeline for small molecules, we present DiffMS, a formula-restricted encoder-decoder generative network that achieves state-of-the-art performance on this task. The encoder utilizes a transformer architecture and models mass spectra domain knowledge such as peak formulae and neutral losses, and the decoder is a discrete graph diffusion model restricted by the heavy-atom composition of a known chemical formula. To develop a robust decoder that bridges latent embeddings and molecular structures, we pretrain the diffusion decoder with fingerprint-structure pairs, which are available in virtually infinite quantities, compared to structure-spectrum pairs that number in the tens of thousands. Extensive experiments on established benchmarks show that DiffMS outperforms existing models on $\textit{de novo}$ molecule generation. We provide several ablations to demonstrate the effectiveness of our diffusion and pretraining approaches and show consistent performance scaling with increasing pretraining dataset size. DiffMS code is publicly available at https://github.com/coleygroup/DiffMS.
Challenging reaction prediction models to generalize to novel chemistry
Jan 11, 2025Deep learning models for anticipating the products of organic reactions have found many use cases, including validating retrosynthetic pathways and constraining synthesis-based molecular design tools. Despite compelling performance on popular benchmark tasks, strange and erroneous predictions sometimes ensue when using these models in practice. The core issue is that common benchmarks test models in an in-distribution setting, whereas many real-world uses for these models are in out-of-distribution settings and require a greater degree of extrapolation. To better understand how current reaction predictors work in out-of-distribution domains, we report a series of more challenging evaluations of a prototypical SMILES-based deep learning model. First, we illustrate how performance on randomly sampled datasets is overly optimistic compared to performance when generalizing to new patents or new authors. Second, we conduct time splits that evaluate how models perform when tested on reactions published in years after those in their training set, mimicking real-world deployment. Finally, we consider extrapolation across reaction classes to reflect what would be required for the discovery of novel reaction types. This panel of tasks can reveal the capabilities and limitations of today's reaction predictors, acting as a crucial first step in the development of tomorrow's next-generation models capable of reaction discovery.
ASKCOS: an open source software suite for synthesis planning
Jan 03, 2025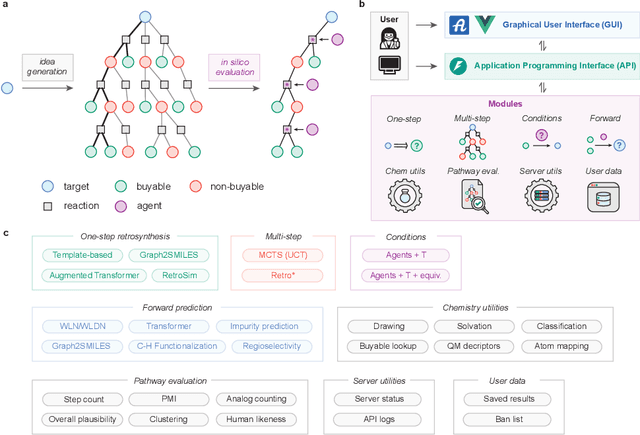



The advancement of machine learning and the availability of large-scale reaction datasets have accelerated the development of data-driven models for computer-aided synthesis planning (CASP) in the past decade. Here, we detail the newest version of ASKCOS, an open source software suite for synthesis planning that makes available several research advances in a freely available, practical tool. Four one-step retrosynthesis models form the basis of both interactive planning and automatic planning modes. Retrosynthetic planning is complemented by other modules for feasibility assessment and pathway evaluation, including reaction condition recommendation, reaction outcome prediction, and auxiliary capabilities such as solubility prediction and quantum mechanical descriptor prediction. ASKCOS has assisted hundreds of medicinal, synthetic, and process chemists in their day-to-day tasks, complementing expert decision making. It is our belief that CASP tools like ASKCOS are an important part of modern chemistry research, and that they offer ever-increasing utility and accessibility.
ShEPhERD: Diffusing shape, electrostatics, and pharmacophores for bioisosteric drug design
Oct 22, 2024Engineering molecules to exhibit precise 3D intermolecular interactions with their environment forms the basis of chemical design. In ligand-based drug design, bioisosteric analogues of known bioactive hits are often identified by virtually screening chemical libraries with shape, electrostatic, and pharmacophore similarity scoring functions. We instead hypothesize that a generative model which learns the joint distribution over 3D molecular structures and their interaction profiles may facilitate 3D interaction-aware chemical design. We specifically design ShEPhERD, an SE(3)-equivariant diffusion model which jointly diffuses/denoises 3D molecular graphs and representations of their shapes, electrostatic potential surfaces, and (directional) pharmacophores to/from Gaussian noise. Inspired by traditional ligand discovery, we compose 3D similarity scoring functions to assess ShEPhERD's ability to conditionally generate novel molecules with desired interaction profiles. We demonstrate ShEPhERD's potential for impact via exemplary drug design tasks including natural product ligand hopping, protein-blind bioactive hit diversification, and bioisosteric fragment merging.
Batched Bayesian optimization with correlated candidate uncertainties
Oct 08, 2024



Batched Bayesian optimization (BO) can accelerate molecular design by efficiently identifying top-performing compounds from a large chemical library. Existing acquisition strategies for batch design in BO aim to balance exploration and exploitation. This often involves optimizing non-additive batch acquisition functions, necessitating approximation via myopic construction and/or diversity heuristics. In this work, we propose an acquisition strategy for discrete optimization that is motivated by pure exploitation, qPO (multipoint Probability of Optimality). qPO maximizes the probability that the batch includes the true optimum, which is expressible as the sum over individual acquisition scores and thereby circumvents the combinatorial challenge of optimizing a batch acquisition function. We differentiate the proposed strategy from parallel Thompson sampling and discuss how it implicitly captures diversity. Finally, we apply our method to the model-guided exploration of large chemical libraries and provide empirical evidence that it performs better than or on par with state-of-the-art methods in batched Bayesian optimization.
Double-Ended Synthesis Planning with Goal-Constrained Bidirectional Search
Jul 08, 2024



Computer-aided synthesis planning (CASP) algorithms have demonstrated expert-level abilities in planning retrosynthetic routes to molecules of low to moderate complexity. However, current search methods assume the sufficiency of reaching arbitrary building blocks, failing to address the common real-world constraint where using specific molecules is desired. To this end, we present a formulation of synthesis planning with starting material constraints. Under this formulation, we propose Double-Ended Synthesis Planning (DESP), a novel CASP algorithm under a bidirectional graph search scheme that interleaves expansions from the target and from the goal starting materials to ensure constraint satisfiability. The search algorithm is guided by a goal-conditioned cost network learned offline from a partially observed hypergraph of valid chemical reactions. We demonstrate the utility of DESP in improving solve rates and reducing the number of search expansions by biasing synthesis planning towards expert goals on multiple new benchmarks. DESP can make use of existing one-step retrosynthesis models, and we anticipate its performance to scale as these one-step model capabilities improve.