 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGenerative structural elucidation from mass spectra as an iterative optimization problem
Feb 07, 2026Liquid chromatography tandem mass spectrometry (LC-MS/MS) is a critical analytical technique for molecular identification across metabolomics, environmental chemistry, and chemical forensics. A variety of computational methods have emerged for structural annotation of spectral features of interest, but many of these features cannot be confidently annotated with reference structures or spectra. Here, we introduce FOAM (Formula-constrained Optimization for Annotating Metabolites), a computational workflow that poses structure elucidation from LC-MS/MS as an iterative optimization problem. FOAM couples a formula-constrained graph genetic algorithm with spectral simulation to explore candidate annotations given an experimental spectrum. We demonstrate FOAM's performance on the NIST'20 and MassSpecGym datasets as both a standalone elucidation pipeline and as a complement to existing inverse models. This work establishes iterative optimization as an effective and extensible paradigm for structural elucidation.
Neural Graph Matching Improves Retrieval Augmented Generation in Molecular Machine Learning
Feb 25, 2025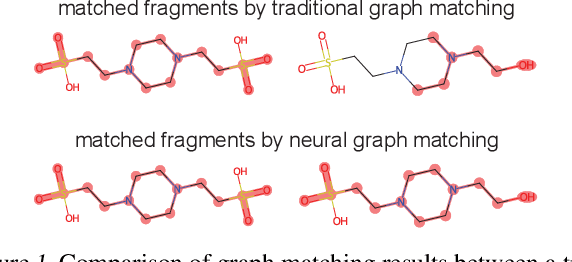
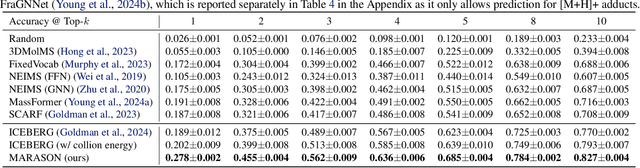
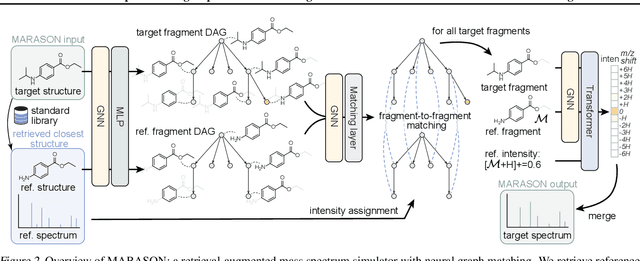
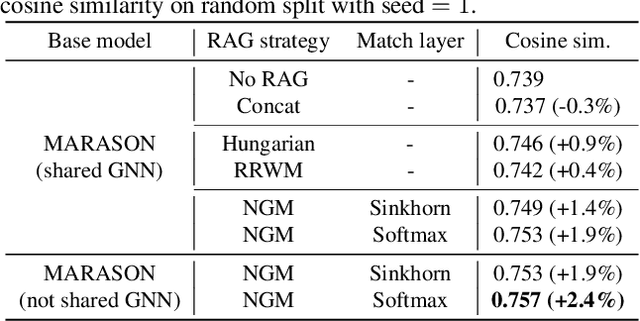
Molecular machine learning has gained popularity with the advancements of geometric deep learning. In parallel, retrieval-augmented generation has become a principled approach commonly used with language models. However, the optimal integration of retrieval augmentation into molecular machine learning remains unclear. Graph neural networks stand to benefit from clever matching to understand the structural alignment of retrieved molecules to a query molecule. Neural graph matching offers a compelling solution by explicitly modeling node and edge affinities between two structural graphs while employing a noise-robust, end-to-end neural network to learn affinity metrics. We apply this approach to mass spectrum simulation and introduce MARASON, a novel model that incorporates neural graph matching to enhance a fragmentation-based neural network. Experimental results highlight the effectiveness of our design, with MARASON achieving 28% top-1 accuracy, a substantial improvement over the non-retrieval state-of-the-art accuracy of 19%. Moreover, MARASON outperforms both naive retrieval-augmented generation methods and traditional graph matching approaches.
DiffMS: Diffusion Generation of Molecules Conditioned on Mass Spectra
Feb 13, 2025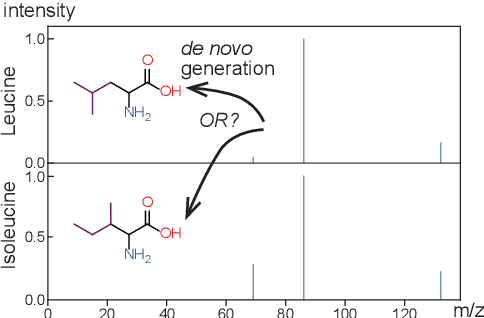
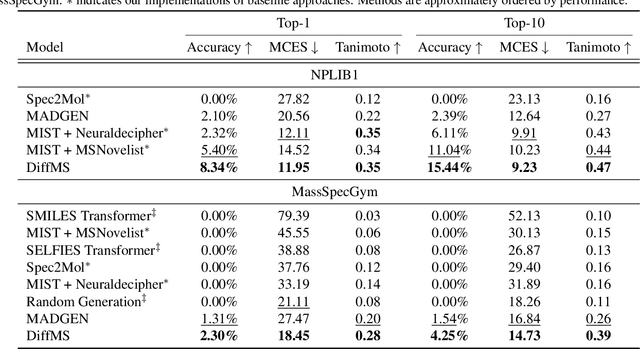
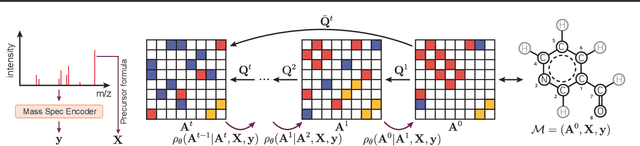
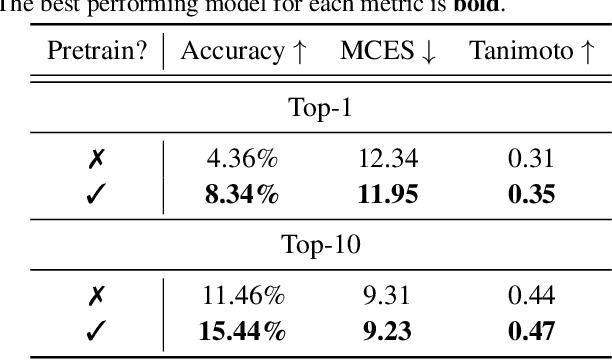
Mass spectrometry plays a fundamental role in elucidating the structures of unknown molecules and subsequent scientific discoveries. One formulation of the structure elucidation task is the conditional $\textit{de novo}$ generation of molecular structure given a mass spectrum. Toward a more accurate and efficient scientific discovery pipeline for small molecules, we present DiffMS, a formula-restricted encoder-decoder generative network that achieves state-of-the-art performance on this task. The encoder utilizes a transformer architecture and models mass spectra domain knowledge such as peak formulae and neutral losses, and the decoder is a discrete graph diffusion model restricted by the heavy-atom composition of a known chemical formula. To develop a robust decoder that bridges latent embeddings and molecular structures, we pretrain the diffusion decoder with fingerprint-structure pairs, which are available in virtually infinite quantities, compared to structure-spectrum pairs that number in the tens of thousands. Extensive experiments on established benchmarks show that DiffMS outperforms existing models on $\textit{de novo}$ molecule generation. We provide several ablations to demonstrate the effectiveness of our diffusion and pretraining approaches and show consistent performance scaling with increasing pretraining dataset size. DiffMS code is publicly available at https://github.com/coleygroup/DiffMS.
Batched Bayesian optimization with correlated candidate uncertainties
Oct 08, 2024



Batched Bayesian optimization (BO) can accelerate molecular design by efficiently identifying top-performing compounds from a large chemical library. Existing acquisition strategies for batch design in BO aim to balance exploration and exploitation. This often involves optimizing non-additive batch acquisition functions, necessitating approximation via myopic construction and/or diversity heuristics. In this work, we propose an acquisition strategy for discrete optimization that is motivated by pure exploitation, qPO (multipoint Probability of Optimality). qPO maximizes the probability that the batch includes the true optimum, which is expressible as the sum over individual acquisition scores and thereby circumvents the combinatorial challenge of optimizing a batch acquisition function. We differentiate the proposed strategy from parallel Thompson sampling and discuss how it implicitly captures diversity. Finally, we apply our method to the model-guided exploration of large chemical libraries and provide empirical evidence that it performs better than or on par with state-of-the-art methods in batched Bayesian optimization.