 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGenerative Model for Synthesizing Ionizable Lipids: A Monte Carlo Tree Search Approach
Dec 01, 2024



Ionizable lipids are essential in developing lipid nanoparticles (LNPs) for effective messenger RNA (mRNA) delivery. While traditional methods for designing new ionizable lipids are typically time-consuming, deep generative models have emerged as a powerful solution, significantly accelerating the molecular discovery process. However, a practical challenge arises as the molecular structures generated can often be difficult or infeasible to synthesize. This project explores Monte Carlo tree search (MCTS)-based generative models for synthesizable ionizable lipids. Leveraging a synthetically accessible lipid building block dataset and two specialized predictors to guide the search through chemical space, we introduce a policy network guided MCTS generative model capable of producing new ionizable lipids with available synthesis pathways.
A Deep Generative Model for the Design of Synthesizable Ionizable Lipids
Dec 01, 2024



Lipid nanoparticles (LNPs) are vital in modern biomedicine, enabling the effective delivery of mRNA for vaccines and therapies by protecting it from rapid degradation. Among the components of LNPs, ionizable lipids play a key role in RNA protection and facilitate its delivery into the cytoplasm. However, designing ionizable lipids is complex. Deep generative models can accelerate this process and explore a larger candidate space compared to traditional methods. Due to the structural differences between lipids and small molecules, existing generative models used for small molecule generation are unsuitable for lipid generation. To address this, we developed a deep generative model specifically tailored for the discovery of ionizable lipids. Our model generates novel ionizable lipid structures and provides synthesis paths using synthetically accessible building blocks, addressing synthesizability. This advancement holds promise for streamlining the development of lipid-based delivery systems, potentially accelerating the deployment of new therapeutic agents, including mRNA vaccines and gene therapies.
Batched Bayesian optimization with correlated candidate uncertainties
Oct 08, 2024



Batched Bayesian optimization (BO) can accelerate molecular design by efficiently identifying top-performing compounds from a large chemical library. Existing acquisition strategies for batch design in BO aim to balance exploration and exploitation. This often involves optimizing non-additive batch acquisition functions, necessitating approximation via myopic construction and/or diversity heuristics. In this work, we propose an acquisition strategy for discrete optimization that is motivated by pure exploitation, qPO (multipoint Probability of Optimality). qPO maximizes the probability that the batch includes the true optimum, which is expressible as the sum over individual acquisition scores and thereby circumvents the combinatorial challenge of optimizing a batch acquisition function. We differentiate the proposed strategy from parallel Thompson sampling and discuss how it implicitly captures diversity. Finally, we apply our method to the model-guided exploration of large chemical libraries and provide empirical evidence that it performs better than or on par with state-of-the-art methods in batched Bayesian optimization.
Diagnosing and fixing common problems in Bayesian optimization for molecule design
Jun 11, 2024



Bayesian optimization (BO) is a principled approach to molecular design tasks. In this paper we explain three pitfalls of BO which can cause poor empirical performance: an incorrect prior width, over-smoothing, and inadequate acquisition function maximization. We show that with these issues addressed, even a basic BO setup is able to achieve the highest overall performance on the PMO benchmark for molecule design (Gao et al, 2022). These results suggest that BO may benefit from more attention in the machine learning for molecules community.
Stochastic Gradient Descent for Gaussian Processes Done Right
Oct 31, 2023



We study the optimisation problem associated with Gaussian process regression using squared loss. The most common approach to this problem is to apply an exact solver, such as conjugate gradient descent, either directly, or to a reduced-order version of the problem. Recently, driven by successes in deep learning, stochastic gradient descent has gained traction as an alternative. In this paper, we show that when done right$\unicode{x2014}$by which we mean using specific insights from the optimisation and kernel communities$\unicode{x2014}$this approach is highly effective. We thus introduce a particular stochastic dual gradient descent algorithm, that may be implemented with a few lines of code using any deep learning framework. We explain our design decisions by illustrating their advantage against alternatives with ablation studies and show that the new method is highly competitive. Our evaluations on standard regression benchmarks and a Bayesian optimisation task set our approach apart from preconditioned conjugate gradients, variational Gaussian process approximations, and a previous version of stochastic gradient descent for Gaussian processes. On a molecular binding affinity prediction task, our method places Gaussian process regression on par in terms of performance with state-of-the-art graph neural networks.
Re-evaluating Retrosynthesis Algorithms with Syntheseus
Oct 30, 2023The planning of how to synthesize molecules, also known as retrosynthesis, has been a growing focus of the machine learning and chemistry communities in recent years. Despite the appearance of steady progress, we argue that imperfect benchmarks and inconsistent comparisons mask systematic shortcomings of existing techniques. To remedy this, we present a benchmarking library called syntheseus which promotes best practice by default, enabling consistent meaningful evaluation of single-step and multi-step retrosynthesis algorithms. We use syntheseus to re-evaluate a number of previous retrosynthesis algorithms, and find that the ranking of state-of-the-art models changes when evaluated carefully. We end with guidance for future works in this area.
Genetic algorithms are strong baselines for molecule generation
Oct 13, 2023Generating molecules, both in a directed and undirected fashion, is a huge part of the drug discovery pipeline. Genetic algorithms (GAs) generate molecules by randomly modifying known molecules. In this paper we show that GAs are very strong algorithms for such tasks, outperforming many complicated machine learning methods: a result which many researchers may find surprising. We therefore propose insisting during peer review that new algorithms must have some clear advantage over GAs, which we call the GA criterion. Ultimately our work suggests that a lot of research in molecule generation should be re-assessed.
Retro-fallback: retrosynthetic planning in an uncertain world
Oct 13, 2023
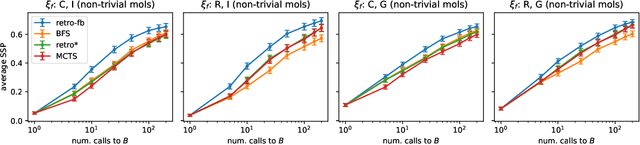


Retrosynthesis is the task of proposing a series of chemical reactions to create a desired molecule from simpler, buyable molecules. While previous works have proposed algorithms to find optimal solutions for a range of metrics (e.g. shortest, lowest-cost), these works generally overlook the fact that we have imperfect knowledge of the space of possible reactions, meaning plans created by the algorithm may not work in a laboratory. In this paper we propose a novel formulation of retrosynthesis in terms of stochastic processes to account for this uncertainty. We then propose a novel greedy algorithm called retro-fallback which maximizes the probability that at least one synthesis plan can be executed in the lab. Using in-silico benchmarks we demonstrate that retro-fallback generally produces better sets of synthesis plans than the popular MCTS and retro* algorithms.
Tanimoto Random Features for Scalable Molecular Machine Learning
Jun 26, 2023The Tanimoto coefficient is commonly used to measure the similarity between molecules represented as discrete fingerprints, either as a distance metric or a positive definite kernel. While many kernel methods can be accelerated using random feature approximations, at present there is a lack of such approximations for the Tanimoto kernel. In this paper we propose two kinds of novel random features to allow this kernel to scale to large datasets, and in the process discover a novel extension of the kernel to real vectors. We theoretically characterize these random features, and provide error bounds on the spectral norm of the Gram matrix. Experimentally, we show that the random features proposed in this work are effective at approximating the Tanimoto coefficient in real-world datasets and that the kernels explored in this work are useful for molecular property prediction and optimization tasks.
Retrosynthetic Planning with Dual Value Networks
Jan 31, 2023Retrosynthesis, which aims to find a route to synthesize a target molecule from commercially available starting materials, is a critical task in drug discovery and materials design. Recently, the combination of ML-based single-step reaction predictors with multi-step planners has led to promising results. However, the single-step predictors are mostly trained offline to optimize the single-step accuracy, without considering complete routes. Here, we leverage reinforcement learning (RL) to improve the single-step predictor, by using a tree-shaped MDP to optimize complete routes while retaining single-step accuracy. Desirable routes should be both synthesizable and of low cost. We propose an online training algorithm, called Planning with Dual Value Networks (PDVN), in which two value networks predict the synthesizability and cost of molecules, respectively. To maintain the single-step accuracy, we design a two-branch network structure for the single-step predictor. On the widely-used USPTO dataset, our PDVN algorithm improves the search success rate of existing multi-step planners (e.g., increasing the success rate from 85.79% to 98.95% for Retro*, and reducing the number of model calls by half while solving 99.47% molecules for RetroGraph). Furthermore, PDVN finds shorter synthesis routes (e.g., reducing the average route length from 5.76 to 4.83 for Retro*, and from 5.63 to 4.78 for RetroGraph).