 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeKL-Regularised Q-Learning: A Token-level Action-Value perspective on Online RLHF
Aug 23, 2025



Proximal Policy Optimisation (PPO) is an established and effective policy gradient algorithm used for Language Model Reinforcement Learning from Human Feedback (LM-RLHF). PPO performs well empirically but has a heuristic motivation and handles the KL-divergence constraint used in LM-RLHF in an ad-hoc manner. In this paper, we develop a a new action-value RL method for the LM-RLHF setting, KL-regularised Q-Learning (KLQ). We then show that our method is equivalent to a version of PPO in a certain specific sense, despite its very different motivation. Finally, we benchmark KLQ on two key language generation tasks -- summarisation and single-turn dialogue. We demonstrate that KLQ performs on-par with PPO at optimising the LM-RLHF objective, and achieves a consistently higher win-rate against PPO on LLM-as-a-judge evaluations.
Tanimoto Random Features for Scalable Molecular Machine Learning
Jun 26, 2023The Tanimoto coefficient is commonly used to measure the similarity between molecules represented as discrete fingerprints, either as a distance metric or a positive definite kernel. While many kernel methods can be accelerated using random feature approximations, at present there is a lack of such approximations for the Tanimoto kernel. In this paper we propose two kinds of novel random features to allow this kernel to scale to large datasets, and in the process discover a novel extension of the kernel to real vectors. We theoretically characterize these random features, and provide error bounds on the spectral norm of the Gram matrix. Experimentally, we show that the random features proposed in this work are effective at approximating the Tanimoto coefficient in real-world datasets and that the kernels explored in this work are useful for molecular property prediction and optimization tasks.
Conditional Neural Processes for Molecules
Oct 17, 2022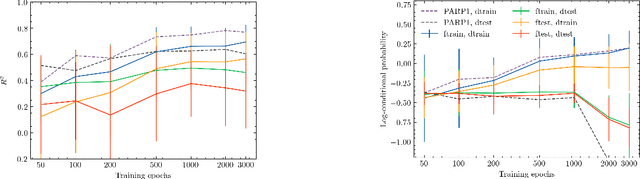
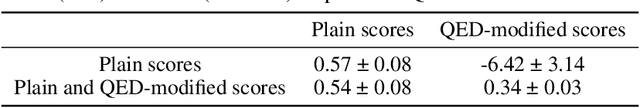
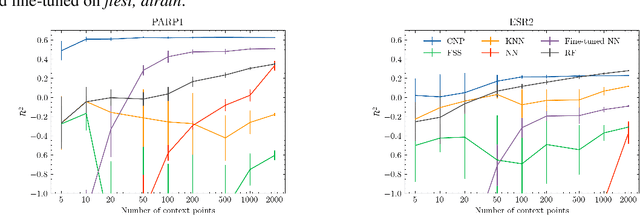
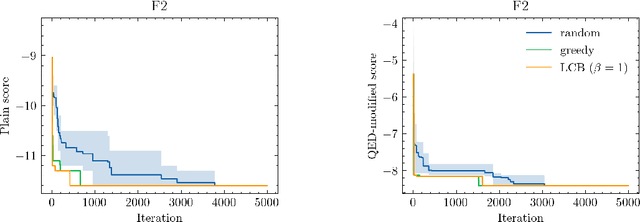
Neural processes (NPs) are models for transfer learning with properties reminiscent of Gaussian Processes (GPs). They are adept at modelling data consisting of few observations of many related functions on the same input space and are trained by minimizing a variational objective, which is computationally much less expensive than the Bayesian updating required by GPs. So far, most studies of NPs have focused on low-dimensional datasets which are not representative of realistic transfer learning tasks. Drug discovery is one application area that is characterized by datasets consisting of many chemical properties or functions which are sparsely observed, yet depend on shared features or representations of the molecular inputs. This paper applies the conditional neural process (CNP) to DOCKSTRING, a dataset of docking scores for benchmarking ML models. CNPs show competitive performance in few-shot learning tasks relative to supervised learning baselines common in QSAR modelling, as well as an alternative model for transfer learning based on pre-training and refining neural network regressors. We present a Bayesian optimization experiment which showcases the probabilistic nature of CNPs and discuss shortcomings of the model in uncertainty quantification.
DOCKSTRING: easy molecular docking yields better benchmarks for ligand design
Oct 29, 2021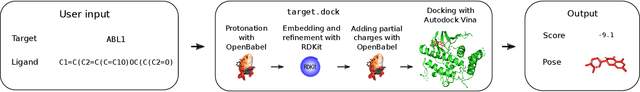
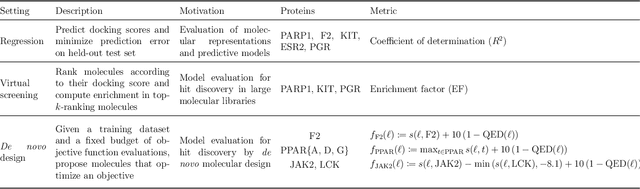
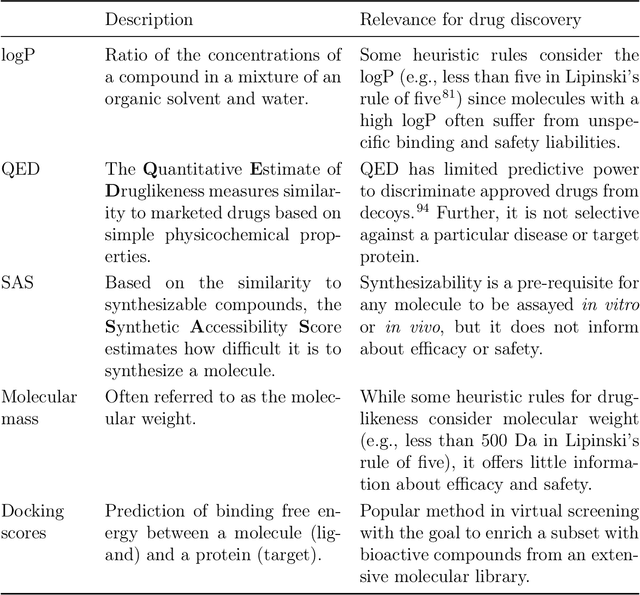
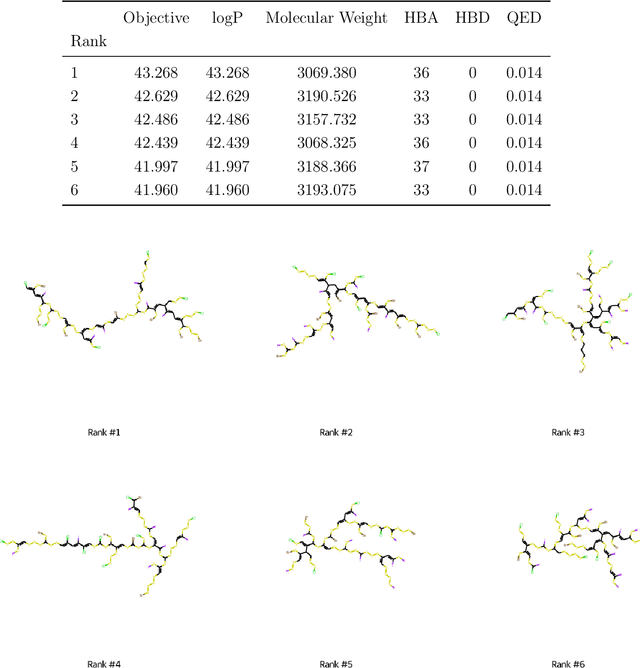
The field of machine learning for drug discovery is witnessing an explosion of novel methods. These methods are often benchmarked on simple physicochemical properties such as solubility or general druglikeness, which can be readily computed. However, these properties are poor representatives of objective functions in drug design, mainly because they do not depend on the candidate's interaction with the target. By contrast, molecular docking is a widely successful method in drug discovery to estimate binding affinities. However, docking simulations require a significant amount of domain knowledge to set up correctly which hampers adoption. To this end, we present DOCKSTRING, a bundle for meaningful and robust comparison of ML models consisting of three components: (1) an open-source Python package for straightforward computation of docking scores; (2) an extensive dataset of docking scores and poses of more than 260K ligands for 58 medically-relevant targets; and (3) a set of pharmaceutically-relevant benchmark tasks including regression, virtual screening, and de novo design. The Python package implements a robust ligand and target preparation protocol that allows non-experts to obtain meaningful docking scores. Our dataset is the first to include docking poses, as well as the first of its size that is a full matrix, thus facilitating experiments in multiobjective optimization and transfer learning. Overall, our results indicate that docking scores are a more appropriate evaluation objective than simple physicochemical properties, yielding more realistic benchmark tasks and molecular candidates.
Sequential Matrix Completion
Oct 23, 2017
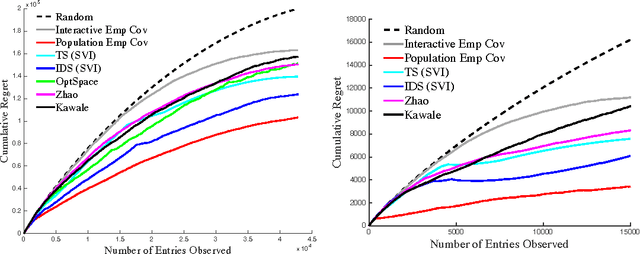
We propose a novel algorithm for sequential matrix completion in a recommender system setting, where the $(i,j)$th entry of the matrix corresponds to a user $i$'s rating of product $j$. The objective of the algorithm is to provide a sequential policy for user-product pair recommendation which will yield the highest possible ratings after a finite time horizon. The algorithm uses a Gamma process factor model with two posterior-focused bandit policies, Thompson Sampling and Information-Directed Sampling. While Thompson Sampling shows competitive performance in simulations, state-of-the-art performance is obtained from Information-Directed Sampling, which makes its recommendations based off a ratio between the expected reward and a measure of information gain. To our knowledge, this is the first implementation of Information Directed Sampling on large real datasets. This approach contributes to a recent line of research on bandit approaches to collaborative filtering including Kawale et al. (2015), Li et al. (2010), Bresler et al. (2014), Li et al. (2016), Deshpande & Montanari (2012), and Zhao et al. (2013). The setting of this paper, as has been noted in Kawale et al. (2015) and Zhao et al. (2013), presents significant challenges to bounding regret after finite horizons. We discuss these challenges in relation to simpler models for bandits with side information, such as linear or gaussian process bandits, and hope the experiments presented here motivate further research toward theoretical guarantees.