 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDOCKSTRING: easy molecular docking yields better benchmarks for ligand design
Paper and Code
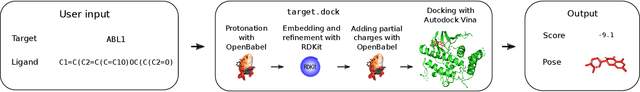
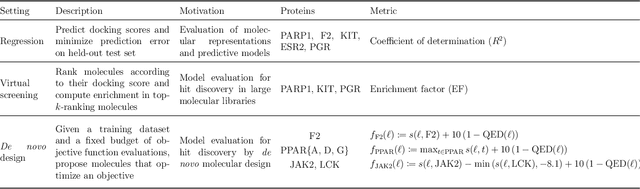
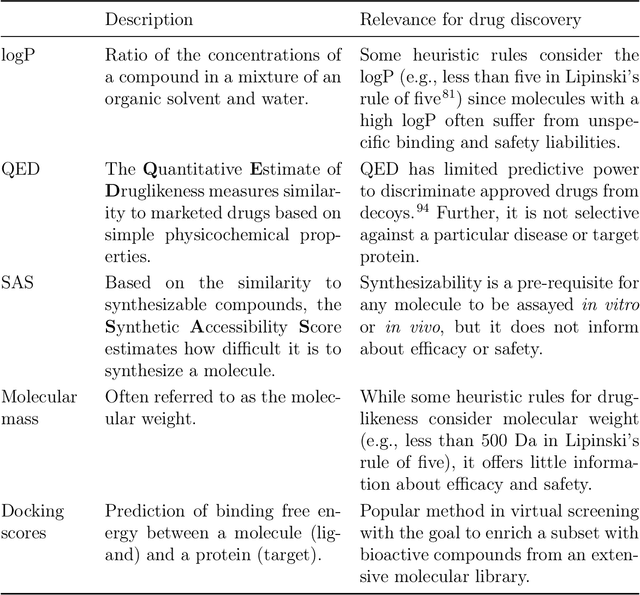
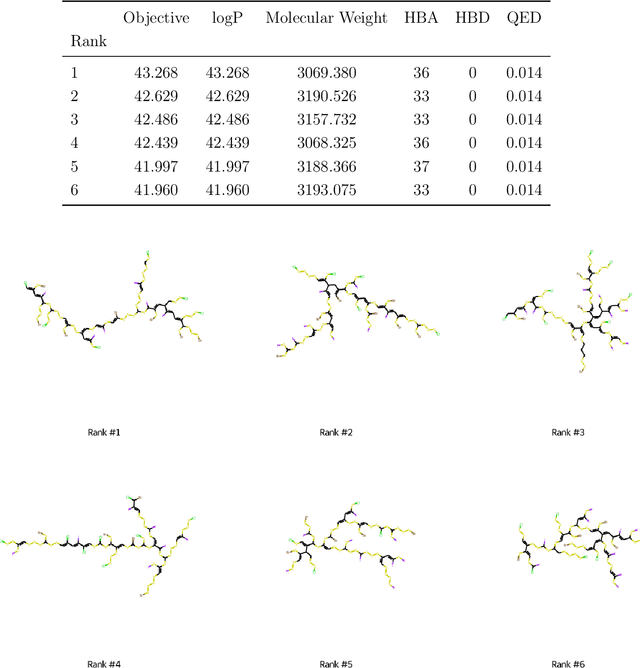
The field of machine learning for drug discovery is witnessing an explosion of novel methods. These methods are often benchmarked on simple physicochemical properties such as solubility or general druglikeness, which can be readily computed. However, these properties are poor representatives of objective functions in drug design, mainly because they do not depend on the candidate's interaction with the target. By contrast, molecular docking is a widely successful method in drug discovery to estimate binding affinities. However, docking simulations require a significant amount of domain knowledge to set up correctly which hampers adoption. To this end, we present DOCKSTRING, a bundle for meaningful and robust comparison of ML models consisting of three components: (1) an open-source Python package for straightforward computation of docking scores; (2) an extensive dataset of docking scores and poses of more than 260K ligands for 58 medically-relevant targets; and (3) a set of pharmaceutically-relevant benchmark tasks including regression, virtual screening, and de novo design. The Python package implements a robust ligand and target preparation protocol that allows non-experts to obtain meaningful docking scores. Our dataset is the first to include docking poses, as well as the first of its size that is a full matrix, thus facilitating experiments in multiobjective optimization and transfer learning. Overall, our results indicate that docking scores are a more appropriate evaluation objective than simple physicochemical properties, yielding more realistic benchmark tasks and molecular candidates.