 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFlow Annealed Importance Sampling Bootstrap
Aug 03, 2022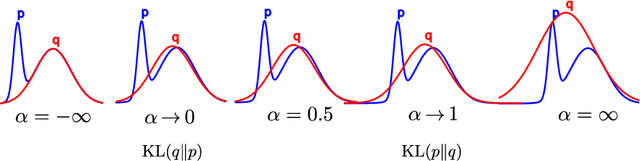
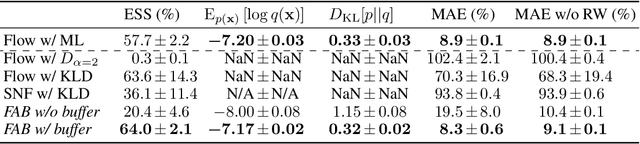
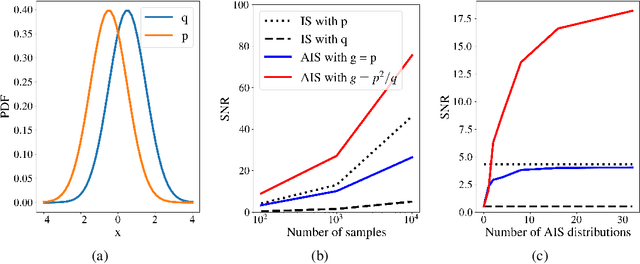

Normalizing flows are tractable density models that can approximate complicated target distributions, e.g. Boltzmann distributions of physical systems. However, current methods for training flows either suffer from mode-seeking behavior, use samples from the target generated beforehand by expensive MCMC simulations, or use stochastic losses that have very high variance. To avoid these problems, we augment flows with annealed importance sampling (AIS) and minimize the mass covering $\alpha$-divergence with $\alpha=2$, which minimizes importance weight variance. Our method, Flow AIS Bootstrap (FAB), uses AIS to generate samples in regions where the flow is a poor approximation of the target, facilitating the discovery of new modes. We target with AIS the minimum variance distribution for the estimation of the $\alpha$-divergence via importance sampling. We also use a prioritized buffer to store and reuse AIS samples. These two features significantly improve FAB's performance. We apply FAB to complex multimodal targets and show that we can approximate them very accurately where previous methods fail. To the best of our knowledge, we are the first to learn the Boltzmann distribution of the alanine dipeptide molecule using only the unnormalized target density and without access to samples generated via Molecular Dynamics (MD) simulations: FAB produces better results than training via maximum likelihood on MD samples while using 100 times fewer target evaluations. After reweighting samples with importance weights, we obtain unbiased histograms of dihedral angles that are almost identical to the ground truth ones.
MACE: Higher Order Equivariant Message Passing Neural Networks for Fast and Accurate Force Fields
Jun 15, 2022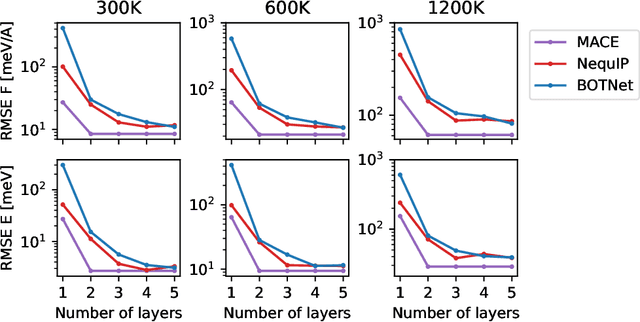
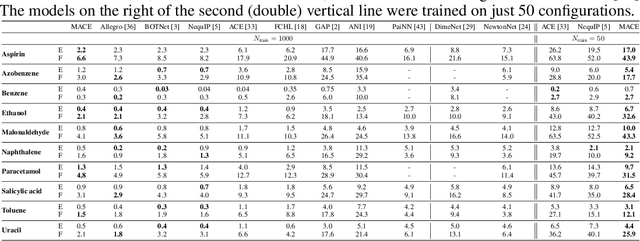

Creating fast and accurate force fields is a long-standing challenge in computational chemistry and materials science. Recently, several equivariant message passing neural networks (MPNNs) have been shown to outperform models built using other approaches in terms of accuracy. However, most MPNNs suffer from high computational cost and poor scalability. We propose that these limitations arise because MPNNs only pass two-body messages leading to a direct relationship between the number of layers and the expressivity of the network. In this work, we introduce MACE, a new equivariant MPNN model that uses higher body order messages. In particular, we show that using four-body messages reduces the required number of message passing iterations to just \emph{two}, resulting in a fast and highly parallelizable model, reaching or exceeding state-of-the-art accuracy on the rMD17, 3BPA, and AcAc benchmark tasks. We also demonstrate that using higher order messages leads to an improved steepness of the learning curves.
The Design Space of E-Equivariant Atom-Centered Interatomic Potentials
May 13, 2022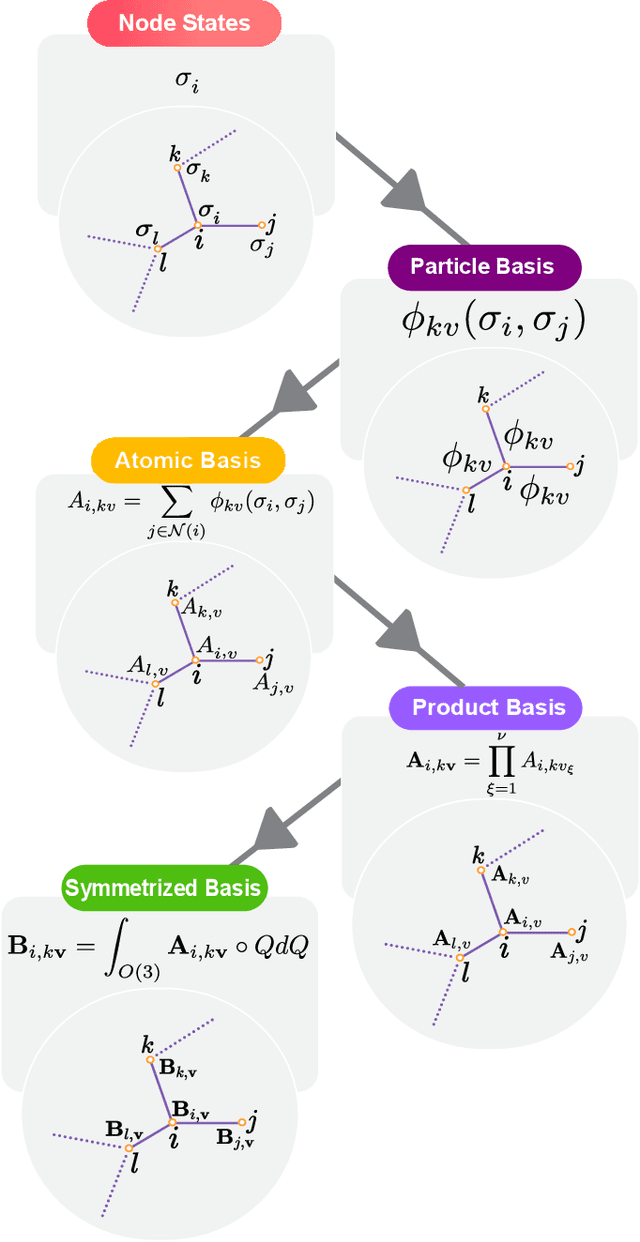
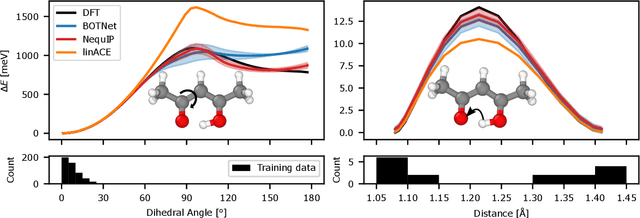
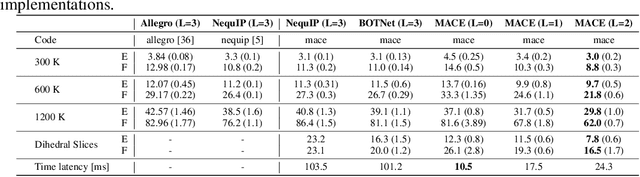
The rapid progress of machine learning interatomic potentials over the past couple of years produced a number of new architectures. Particularly notable among these are the Atomic Cluster Expansion (ACE), which unified many of the earlier ideas around atom density-based descriptors, and Neural Equivariant Interatomic Potentials (NequIP), a message passing neural network with equivariant features that showed state of the art accuracy. In this work, we construct a mathematical framework that unifies these models: ACE is generalised so that it can be recast as one layer of a multi-layer architecture. From another point of view, the linearised version of NequIP is understood as a particular sparsification of a much larger polynomial model. Our framework also provides a practical tool for systematically probing different choices in the unified design space. We demonstrate this by an ablation study of NequIP via a set of experiments looking at in- and out-of-domain accuracy and smooth extrapolation very far from the training data, and shed some light on which design choices are critical for achieving high accuracy. Finally, we present BOTNet (Body-Ordered-Tensor-Network), a much-simplified version of NequIP, which has an interpretable architecture and maintains accuracy on benchmark datasets.
Bootstrap Your Flow
Dec 06, 2021
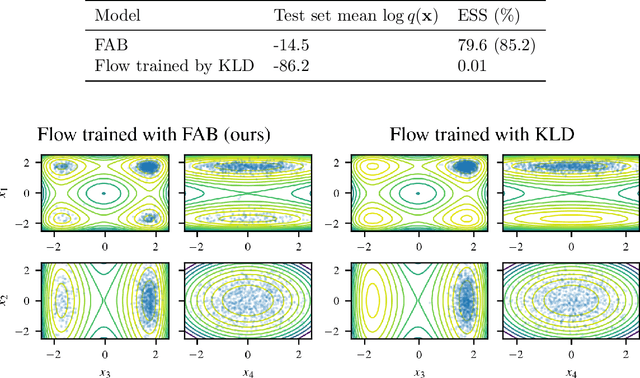
Normalising flows are flexible, parameterized distributions that can be used to approximate expectations from intractable distributions via importance sampling. However, current flow-based approaches are limited on challenging targets where they either suffer from mode seeking behaviour or high variance in the training loss, or rely on samples from the target distribution, which may not be available. To address these challenges, we combine flows with annealed importance sampling (AIS), while using the $\alpha$-divergence as our objective, in a novel training procedure, FAB (Flow AIS Bootstrap). Thereby, the flow and AIS to improve each other in a bootstrapping manner. We demonstrate that FAB can be used to produce accurate approximations to complex target distributions, including Boltzmann distributions, in problems where previous flow-based methods fail.
DOCKSTRING: easy molecular docking yields better benchmarks for ligand design
Oct 29, 2021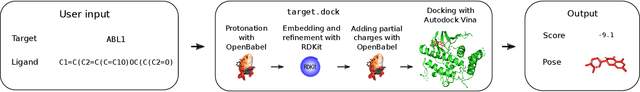
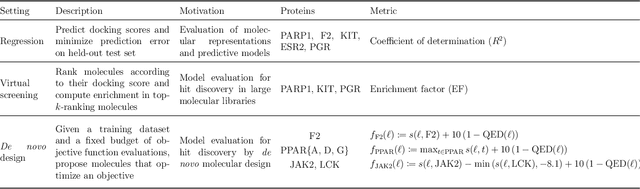
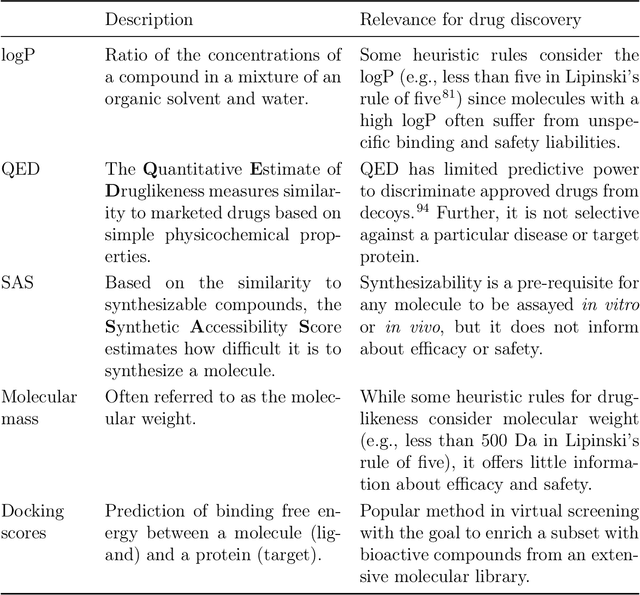
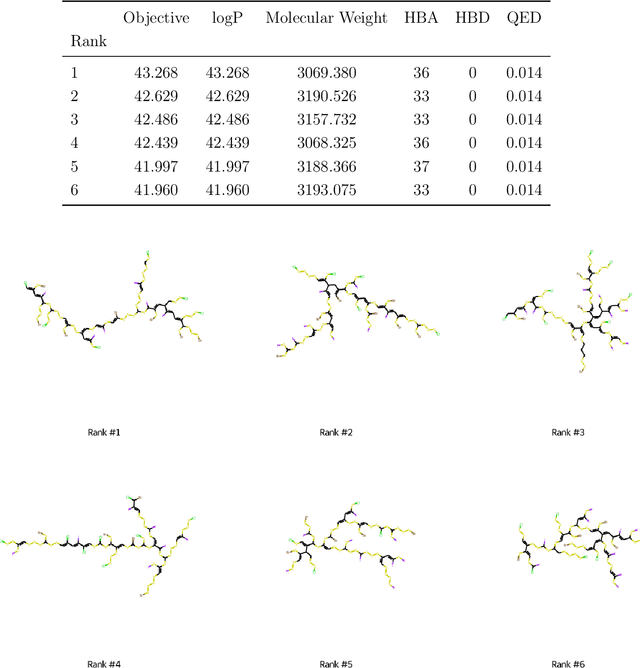
The field of machine learning for drug discovery is witnessing an explosion of novel methods. These methods are often benchmarked on simple physicochemical properties such as solubility or general druglikeness, which can be readily computed. However, these properties are poor representatives of objective functions in drug design, mainly because they do not depend on the candidate's interaction with the target. By contrast, molecular docking is a widely successful method in drug discovery to estimate binding affinities. However, docking simulations require a significant amount of domain knowledge to set up correctly which hampers adoption. To this end, we present DOCKSTRING, a bundle for meaningful and robust comparison of ML models consisting of three components: (1) an open-source Python package for straightforward computation of docking scores; (2) an extensive dataset of docking scores and poses of more than 260K ligands for 58 medically-relevant targets; and (3) a set of pharmaceutically-relevant benchmark tasks including regression, virtual screening, and de novo design. The Python package implements a robust ligand and target preparation protocol that allows non-experts to obtain meaningful docking scores. Our dataset is the first to include docking poses, as well as the first of its size that is a full matrix, thus facilitating experiments in multiobjective optimization and transfer learning. Overall, our results indicate that docking scores are a more appropriate evaluation objective than simple physicochemical properties, yielding more realistic benchmark tasks and molecular candidates.
Symmetry-Aware Actor-Critic for 3D Molecular Design
Nov 25, 2020
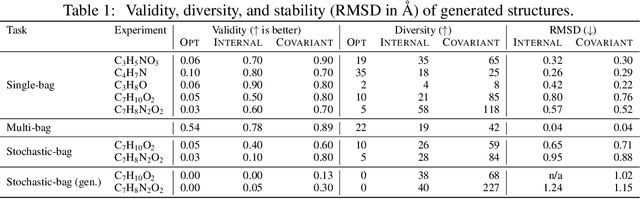


Automating molecular design using deep reinforcement learning (RL) has the potential to greatly accelerate the search for novel materials. Despite recent progress on leveraging graph representations to design molecules, such methods are fundamentally limited by the lack of three-dimensional (3D) information. In light of this, we propose a novel actor-critic architecture for 3D molecular design that can generate molecular structures unattainable with previous approaches. This is achieved by exploiting the symmetries of the design process through a rotationally covariant state-action representation based on a spherical harmonics series expansion. We demonstrate the benefits of our approach on several 3D molecular design tasks, where we find that building in such symmetries significantly improves generalization and the quality of generated molecules.
Reinforcement Learning for Molecular Design Guided by Quantum Mechanics
Feb 18, 2020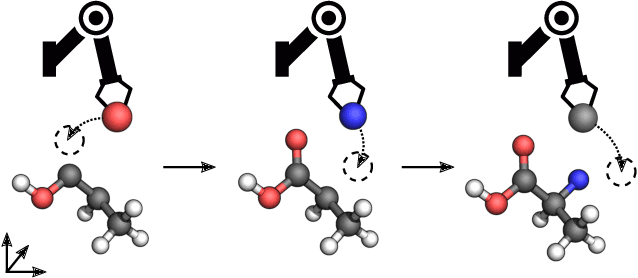


Automating molecular design using deep reinforcement learning (RL) holds the promise of accelerating the discovery of new chemical compounds. A limitation of existing approaches is that they work with molecular graphs and thus ignore the location of atoms in space, which restricts them to 1) generating single organic molecules and 2) heuristic reward functions. To address this, we present a novel RL formulation for molecular design in Cartesian coordinates, thereby extending the class of molecules that can be built. Our reward function is directly based on fundamental physical properties such as the energy, which we approximate via fast quantum-chemical methods. To enable progress towards de-novo molecular design, we introduce MolGym, an RL environment comprising several challenging molecular design tasks along with baselines. In our experiments, we show that our agent can efficiently learn to solve these tasks from scratch by working in a translation and rotation invariant state-action space.
A Generative Model for Molecular Distance Geometry
Oct 03, 2019


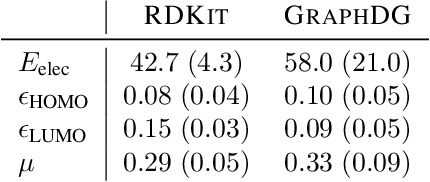
Computing equilibrium states for many-body systems, such as molecules, is a long-standing challenge. In the absence of methods for generating statistically independent samples, great computational effort is invested in simulating these systems using, for example, Markov chain Monte Carlo. We present a probabilistic model that generates such samples for molecules from their graph representations. Our model learns a low-dimensional manifold that preserves the geometry of local atomic neighborhoods through a principled learning representation that is based on Euclidean distance geometry. We create a new dataset for molecular conformation generation with which we show experimentally that our generative model achieves state-of-the-art accuracy. Finally, we show how to use our model as a proposal distribution in an importance sampling scheme to compute molecular properties.