 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAI-Driven Expansion and Application of the Alexandria Database
Dec 09, 2025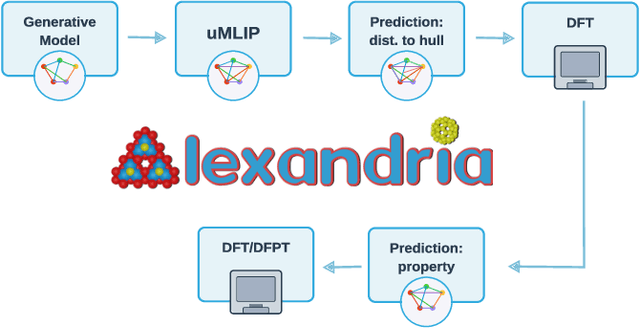
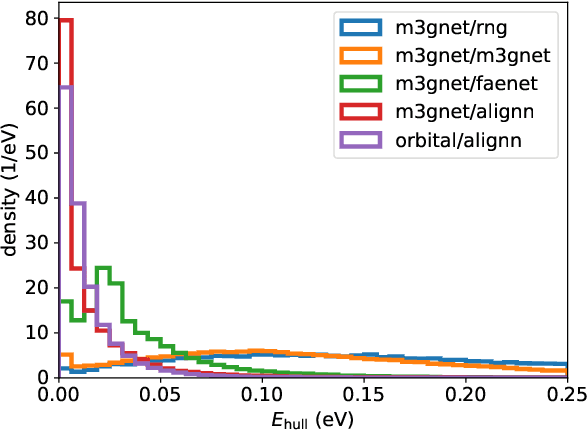
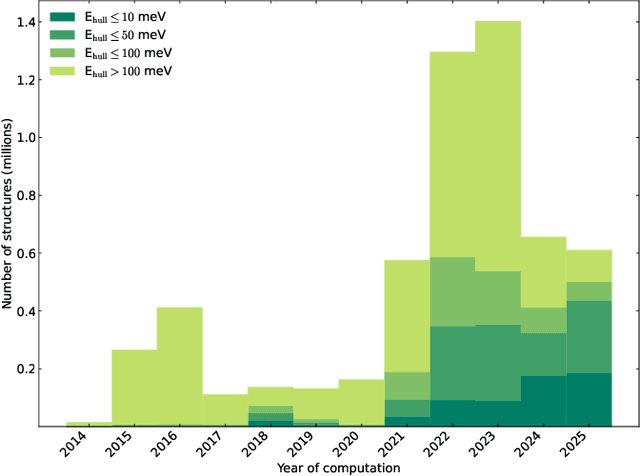
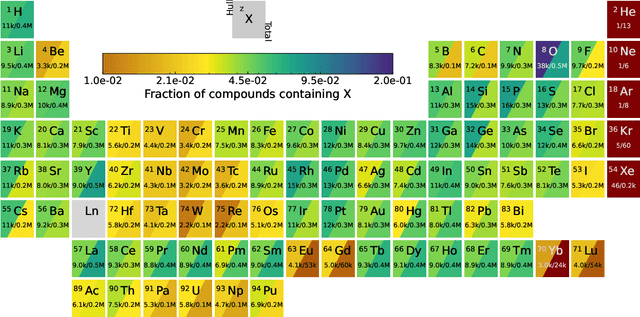
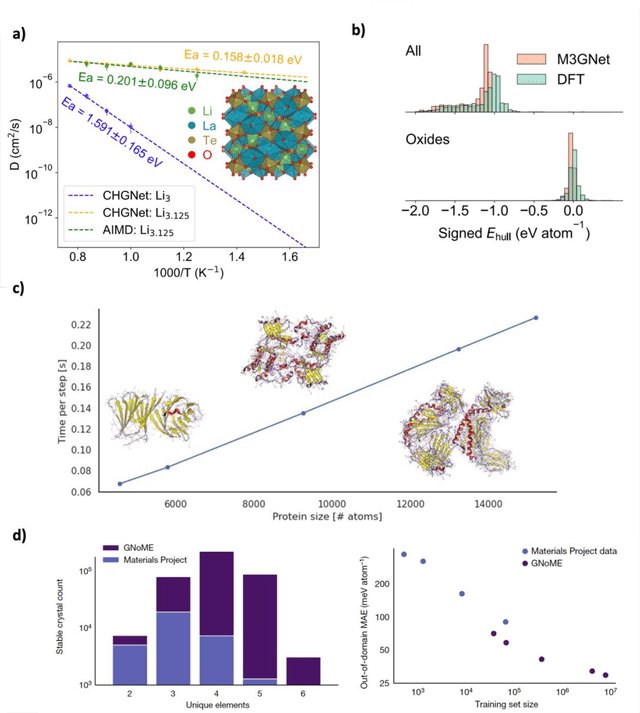
We present a novel multi-stage workflow for computational materials discovery that achieves a 99% success rate in identifying compounds within 100 meV/atom of thermodynamic stability, with a threefold improvement over previous approaches. By combining the Matra-Genoa generative model, Orb-v2 universal machine learning interatomic potential, and ALIGNN graph neural network for energy prediction, we generated 119 million candidate structures and added 1.3 million DFT-validated compounds to the ALEXANDRIA database, including 74 thousand new stable materials. The expanded ALEXANDRIA database now contains 5.8 million structures with 175 thousand compounds on the convex hull. Predicted structural disorder rates (37-43%) match experimental databases, unlike other recent AI-generated datasets. Analysis reveals fundamental patterns in space group distributions, coordination environments, and phase stability networks, including sub-linear scaling of convex hull connectivity. We release the complete dataset, including sAlex25 with 14 million out-of-equilibrium structures containing forces and stresses for training universal force fields. We demonstrate that fine-tuning a GRACE model on this data improves benchmark accuracy. All data, models, and workflows are freely available under Creative Commons licenses.
A practical guide to machine learning interatomic potentials -- Status and future
Mar 12, 2025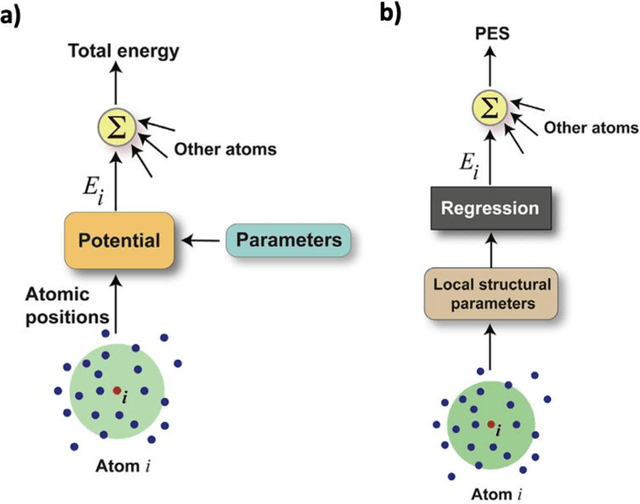

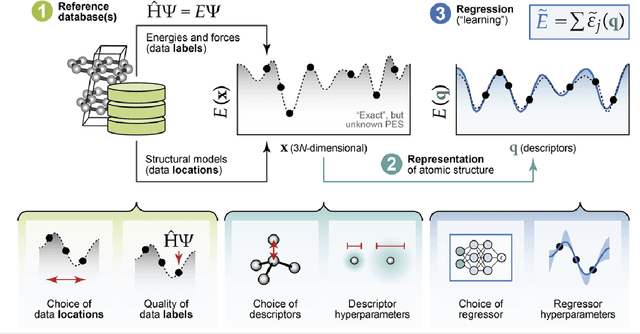
The rapid development and large body of literature on machine learning interatomic potentials (MLIPs) can make it difficult to know how to proceed for researchers who are not experts but wish to use these tools. The spirit of this review is to help such researchers by serving as a practical, accessible guide to the state-of-the-art in MLIPs. This review paper covers a broad range of topics related to MLIPs, including (i) central aspects of how and why MLIPs are enablers of many exciting advancements in molecular modeling, (ii) the main underpinnings of different types of MLIPs, including their basic structure and formalism, (iii) the potentially transformative impact of universal MLIPs for both organic and inorganic systems, including an overview of the most recent advances, capabilities, downsides, and potential applications of this nascent class of MLIPs, (iv) a practical guide for estimating and understanding the execution speed of MLIPs, including guidance for users based on hardware availability, type of MLIP used, and prospective simulation size and time, (v) a manual for what MLIP a user should choose for a given application by considering hardware resources, speed requirements, energy and force accuracy requirements, as well as guidance for choosing pre-trained potentials or fitting a new potential from scratch, (vi) discussion around MLIP infrastructure, including sources of training data, pre-trained potentials, and hardware resources for training, (vii) summary of some key limitations of present MLIPs and current approaches to mitigate such limitations, including methods of including long-range interactions, handling magnetic systems, and treatment of excited states, and finally (viii) we finish with some more speculative thoughts on what the future holds for the development and application of MLIPs over the next 3-10+ years.
The Design Space of E-Equivariant Atom-Centered Interatomic Potentials
May 13, 2022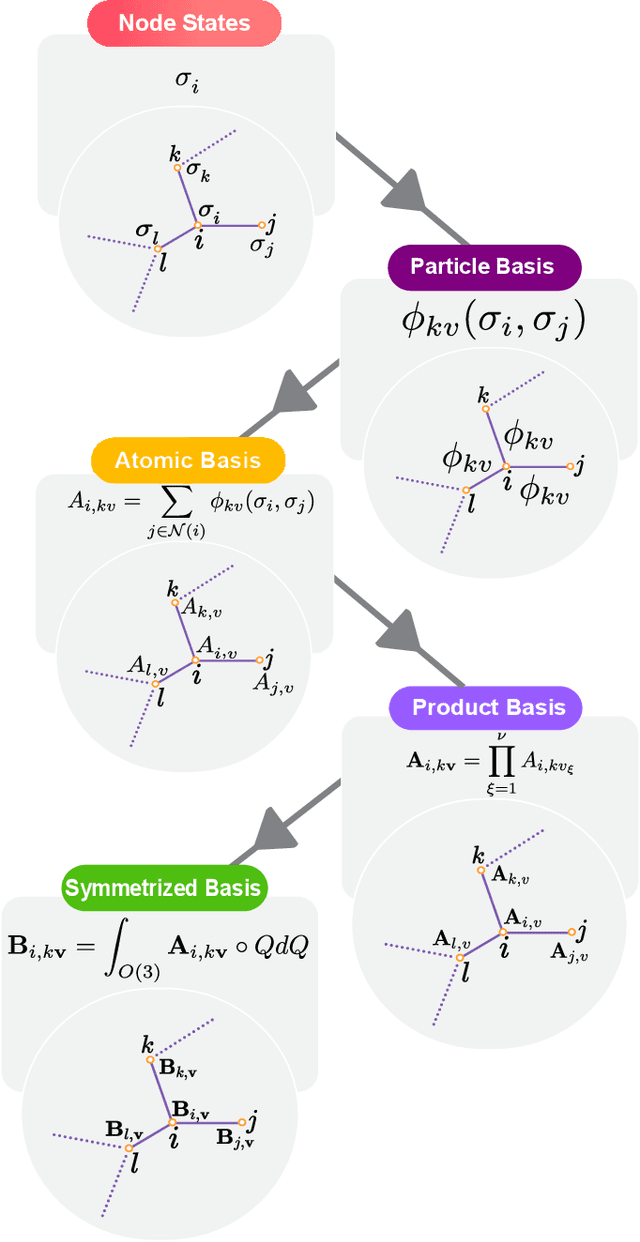
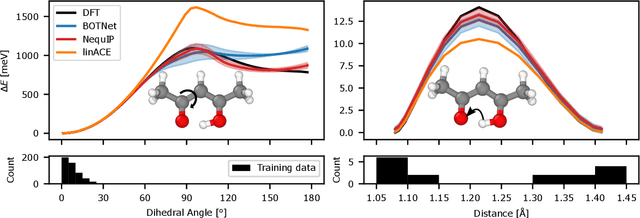
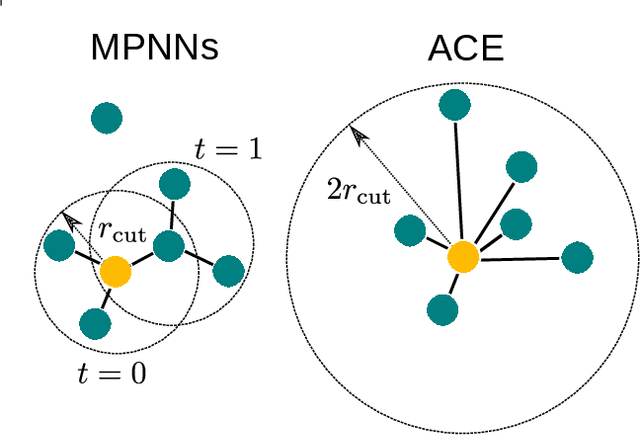
The rapid progress of machine learning interatomic potentials over the past couple of years produced a number of new architectures. Particularly notable among these are the Atomic Cluster Expansion (ACE), which unified many of the earlier ideas around atom density-based descriptors, and Neural Equivariant Interatomic Potentials (NequIP), a message passing neural network with equivariant features that showed state of the art accuracy. In this work, we construct a mathematical framework that unifies these models: ACE is generalised so that it can be recast as one layer of a multi-layer architecture. From another point of view, the linearised version of NequIP is understood as a particular sparsification of a much larger polynomial model. Our framework also provides a practical tool for systematically probing different choices in the unified design space. We demonstrate this by an ablation study of NequIP via a set of experiments looking at in- and out-of-domain accuracy and smooth extrapolation very far from the training data, and shed some light on which design choices are critical for achieving high accuracy. Finally, we present BOTNet (Body-Ordered-Tensor-Network), a much-simplified version of NequIP, which has an interpretable architecture and maintains accuracy on benchmark datasets.