 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDesign Space of Self--Consistent Electrostatic Machine Learning Interatomic Potentials
Mar 16, 2026Machine learning interatomic potentials (MLIPs) have become widely used tools in atomistic simulations. For much of the history of this field, the most commonly employed architectures were based on short-ranged atomic energy contributions, and the assumption of locality still persists in many modern foundation models. While this approach has enabled efficient and accurate modelling for many use cases, it poses intrinsic limitations for systems where long-range electrostatics, charge transfer, or induced polarization play a central role. A growing body of work has proposed extensions that incorporate electrostatic effects, ranging from locally predicted atomic charges to self-consistent models. While these models have demonstrated success for specific examples, their underlying assumptions, and fundamental limitations are not yet well understood. In this work, we present a framework for treating electrostatics in MLIPs by viewing existing models as coarse-grained approximations to density functional theory (DFT). This perspective makes explicit the approximations involved, clarifies the physical meaning of the learned quantities, and reveals connections and equivalences between several previously proposed models. Using this formalism, we identify key design choices that define a broader design space of self-consistent electrostatic MLIPs. We implement salient points in this space using the MACE architecture and a shared representation of the charge density, enabling controlled comparisons between different approaches. Finally, we evaluate these models on two instructive test cases: metal-water interfaces, which probe the contrasting electrostatic response of conducting and insulating systems, and charged vacancies in silicon dioxide. Our results highlight the limitations of existing approaches and demonstrate how more expressive self-consistent models are needed to resolve failures.
MACE-POLAR-1: A Polarisable Electrostatic Foundation Model for Molecular Chemistry
Feb 23, 2026Accurate modelling of electrostatic interactions and charge transfer is fundamental to computational chemistry, yet most machine learning interatomic potentials (MLIPs) rely on local atomic descriptors that cannot capture long-range electrostatic effects. We present a new electrostatic foundation model for molecular chemistry that extends the MACE architecture with explicit treatment of long-range interactions and electrostatic induction. Our approach combines local many-body geometric features with a non-self-consistent field formalism that updates learnable charge and spin densities through polarisable iterations to model induction, followed by global charge equilibration via learnable Fukui functions to control total charge and total spin. This design enables an accurate and physical description of systems with varying charge and spin states while maintaining computational efficiency. Trained on the OMol25 dataset of 100 million hybrid DFT calculations, our models achieve chemical accuracy across diverse benchmarks, with accuracy competitive with hybrid DFT on thermochemistry, reaction barriers, conformational energies, and transition metal complexes. Notably, we demonstrate that the inclusion of long-range electrostatics leads to a large improvement in the description of non-covalent interactions and supramolecular complexes over non-electrostatic models, including sub-kcal/mol prediction of molecular crystal formation energy in the X23-DMC dataset and a fourfold improvement over short-ranged models on protein-ligand interactions. The model's ability to handle variable charge and spin states, respond to external fields, provide interpretable spin-resolved charge densities, and maintain accuracy from small molecules to protein-ligand complexes positions it as a versatile tool for computational molecular chemistry and drug discovery.
Better without U: Impact of Selective Hubbard U Correction on Foundational MLIPs
Jan 28, 2026The training of foundational machine learning interatomic potentials (fMLIPs) relies on diverse databases with energies and forces calculated using ab initio methods. We show that fMLIPs trained on large datasets such as MPtrj, Alexandria, and OMat24 encode inconsistencies from the Materials Project's selective use of the Hubbard U correction, which is applied to certain transition metals only if O or F atoms are present in the simulation cell. This inconsistent use of +U creates two incompatible potential-energy surfaces (PES): a lower-energy GGA surface and a higher-energy GGA+U one. When trained on both, MLIPs interpolate between them, leading to systematic underbinding, or even spurious repulsion, between U-corrected metals and oxygen- or fluorine-containing species. Models such as MACE-OMAT and -MPA exhibit repulsion between U-corrected metals and their oxides, limiting their value for studying catalysis and oxidation. We link the severity of this pathology to the oxygen number density in U-corrected training configurations. This explains why OMAT-trained models are most affected and suggests the issue might worsen as expanding future datasets increasingly include configurations with low oxygen content, such as those generated through combinatorial exploration of multi-element or defect-containing systems. Our simple per-U-corrected-atom shift aligns PBE+U and PBE energies for identical structures, yielding a smoother PES compared to existing correction schemes, which target phase diagram accuracy. As a result, models trained on datasets with our shift applied exhibit smaller mean absolute errors for the adsorption energies of oxygen on U-corrected elemental slabs. Since datasets omitting +U entirely (e.g. MatPES, MP-ALOE) avoid these pathologies, we recommend excluding +U in future fMLIP datasets. For existing datasets, our post-hoc correction provides a low-cost improvement.
Resonances in reflective Hamiltonian Monte Carlo
Apr 16, 2025



In high dimensions, reflective Hamiltonian Monte Carlo with inexact reflections exhibits slow mixing when the particle ensemble is initialised from a Dirac delta distribution and the uniform distribution is targeted. By quantifying the instantaneous non-uniformity of the distribution with the Sinkhorn divergence, we elucidate the principal mechanisms underlying the mixing problems. In spheres and cubes, we show that the collective motion transitions between fluid-like and discretisation-dominated behaviour, with the critical step size scaling as a power law in the dimension. In both regimes, the particles can spontaneously unmix, leading to resonances in the particle density and the aforementioned problems. Additionally, low-dimensional toy models of the dynamics are constructed which reproduce the dominant features of the high-dimensional problem. Finally, the dynamics is contrasted with the exact Hamiltonian particle flow and tuning practices are discussed.
Accurate Crystal Structure Prediction of New 2D Hybrid Organic Inorganic Perovskites
Mar 11, 2024Low dimensional hybrid organic-inorganic perovskites (HOIPs) represent a promising class of electronically active materials for both light absorption and emission. The design space of HOIPs is extremely large, since a diverse space of organic cations can be combined with different inorganic frameworks. This immense design space allows for tunable electronic and mechanical properties, but also necessitates the development of new tools for in silico high throughput analysis of candidate structures. In this work, we present an accurate, efficient, transferable and widely applicable machine learning interatomic potential (MLIP) for predicting the structure of new 2D HOIPs. Using the MACE architecture, an MLIP is trained on 86 diverse experimentally reported HOIP structures. The model is tested on 73 unseen perovskite compositions, and achieves chemical accuracy with respect to the reference electronic structure method. Our model is then combined with a simple random structure search algorithm to predict the structure of hypothetical HOIPs given only the proposed composition. Success is demonstrated by correctly and reliably recovering the crystal structure of a set of experimentally known 2D perovskites. Such a random structure search is impossible with ab initio methods due to the associated computational cost, but is relatively inexpensive with the MACE potential. Finally, the procedure is used to predict the structure formed by a new organic cation with no previously known corresponding perovskite. Laboratory synthesis of the new hybrid perovskite confirms the accuracy of our prediction. This capability, applied at scale, enables efficient screening of thousands of combinations of organic cations and inorganic layers.
Zero Shot Molecular Generation via Similarity Kernels
Feb 13, 2024



Generative modelling aims to accelerate the discovery of novel chemicals by directly proposing structures with desirable properties. Recently, score-based, or diffusion, generative models have significantly outperformed previous approaches. Key to their success is the close relationship between the score and physical force, allowing the use of powerful equivariant neural networks. However, the behaviour of the learnt score is not yet well understood. Here, we analyse the score by training an energy-based diffusion model for molecular generation. We find that during the generation the score resembles a restorative potential initially and a quantum-mechanical force at the end. In between the two endpoints, it exhibits special properties that enable the building of large molecules. Using insights from the trained model, we present Similarity-based Molecular Generation (SiMGen), a new method for zero shot molecular generation. SiMGen combines a time-dependent similarity kernel with descriptors from a pretrained machine learning force field to generate molecules without any further training. Our approach allows full control over the molecular shape through point cloud priors and supports conditional generation. We also release an interactive web tool that allows users to generate structures with SiMGen online (https://zndraw.icp.uni-stuttgart.de).
Energy-conserving equivariant GNN for elasticity of lattice architected metamaterials
Jan 30, 2024



Lattices are architected metamaterials whose properties strongly depend on their geometrical design. The analogy between lattices and graphs enables the use of graph neural networks (GNNs) as a faster surrogate model compared to traditional methods such as finite element modelling. In this work we present a higher-order GNN model trained to predict the fourth-order stiffness tensor of periodic strut-based lattices. The key features of the model are (i) SE(3) equivariance, and (ii) consistency with the thermodynamic law of conservation of energy. We compare the model to non-equivariant models based on a number of error metrics and demonstrate the benefits of the encoded equivariance and energy conservation in terms of predictive performance and reduced training requirements.
Equivariant Matrix Function Neural Networks
Oct 16, 2023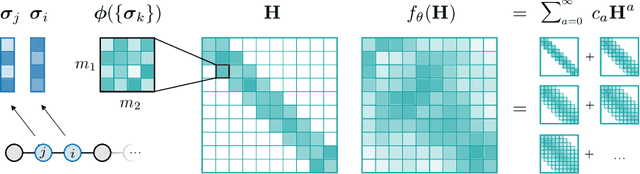
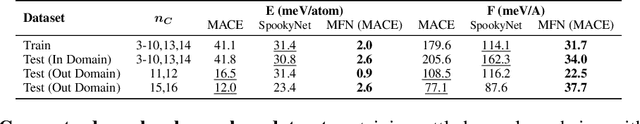
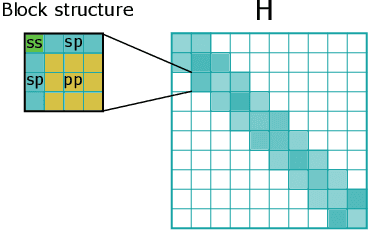

Graph Neural Networks (GNNs), especially message-passing neural networks (MPNNs), have emerged as powerful architectures for learning on graphs in diverse applications. However, MPNNs face challenges when modeling non-local interactions in systems such as large conjugated molecules, metals, or amorphous materials. Although Spectral GNNs and traditional neural networks such as recurrent neural networks and transformers mitigate these challenges, they often lack extensivity, adaptability, generalizability, computational efficiency, or fail to capture detailed structural relationships or symmetries in the data. To address these concerns, we introduce Matrix Function Neural Networks (MFNs), a novel architecture that parameterizes non-local interactions through analytic matrix equivariant functions. Employing resolvent expansions offers a straightforward implementation and the potential for linear scaling with system size. The MFN architecture achieves state-of-the-art performance in standard graph benchmarks, such as the ZINC and TU datasets, and is able to capture intricate non-local interactions in quantum systems, paving the way to new state-of-the-art force fields.
Hyperactive Learning (HAL) for Data-Driven Interatomic Potentials
Oct 09, 2022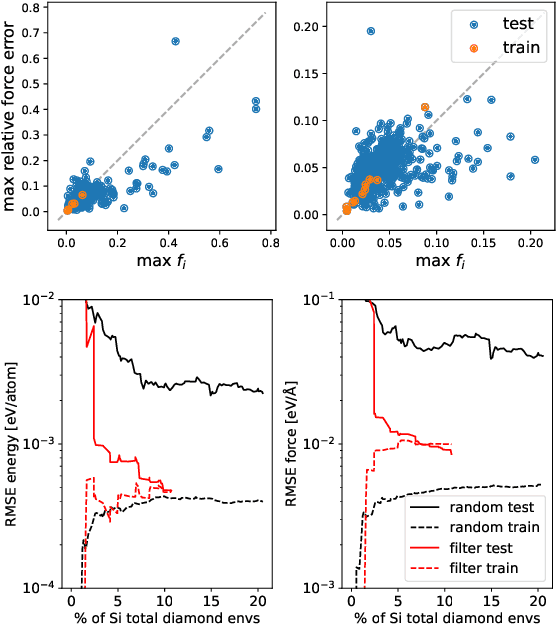
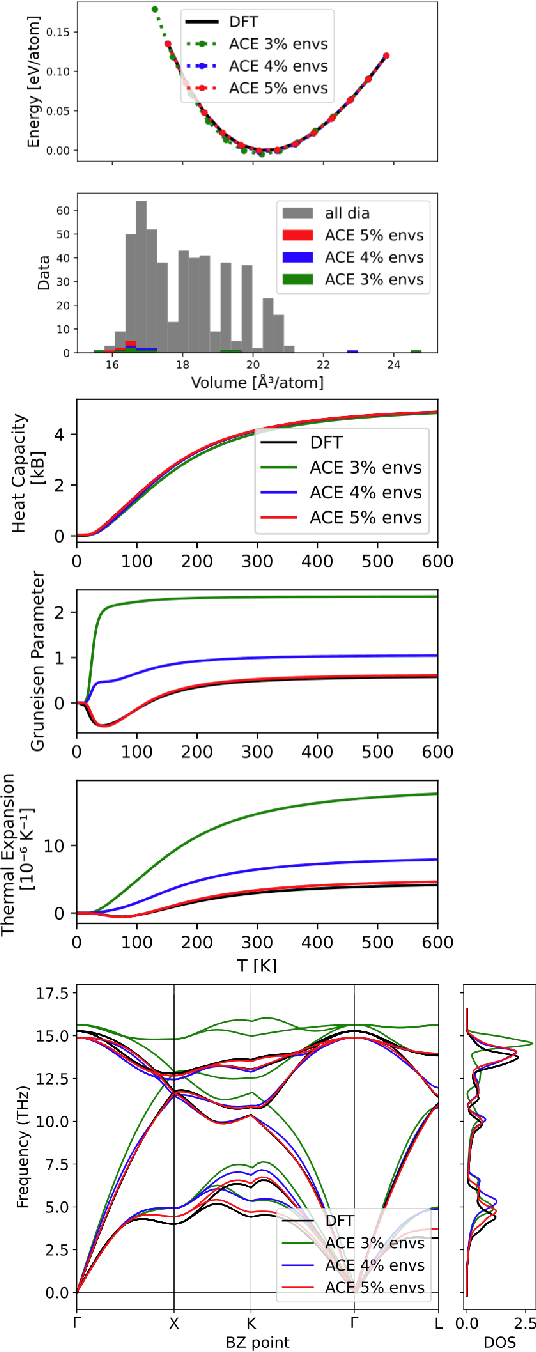
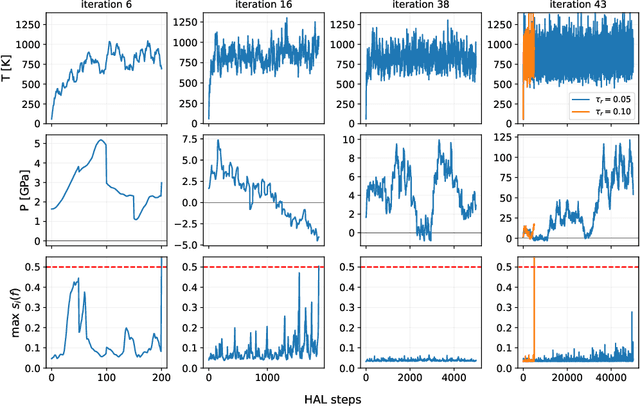
Data-driven interatomic potentials have emerged as a powerful class of surrogate models for ab initio potential energy surfaces that are able to reliably predict macroscopic properties with experimental accuracy. In generating accurate and transferable potentials the most time-consuming and arguably most important task is generating the training set, which still requires significant expert user input. To accelerate this process, this work presents hyperactive learning (HAL), a framework for formulating an accelerated sampling algorithm specifically for the task of training database generation. The overarching idea is to start from a physically motivated sampler (e.g., molecular dynamics) and a biasing term that drives the system towards high uncertainty and thus to unseen training configurations. Building on this framework, general protocols for building training databases for alloys and polymers leveraging the HAL framework will be presented. For alloys, fast (< 100 microsecond/atom/cpu-core) ACE potentials for AlSi10 are created that able to predict the melting temperature with good accuracy by fitting to a minimal HAL-generated database containing 88 configurations (32 atoms each) in 17 seconds using 8 cpu threads. For polymers, a HAL database is built using ACE able to determine the density of a long polyethylene glycol (PEG) polymer formed of 200 monomer units with experimental accuracy by only fitting to small isolated PEG polymers with sizes ranging from 2 to 32.
Tensor-reduced atomic density representations
Oct 02, 2022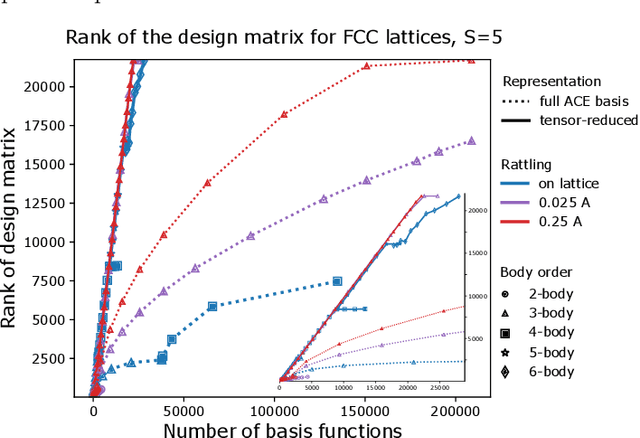
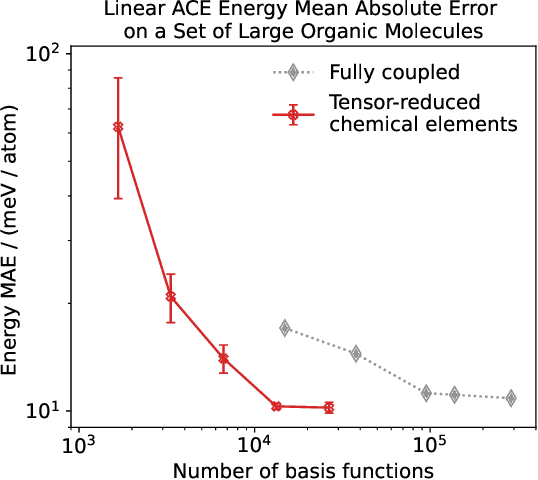
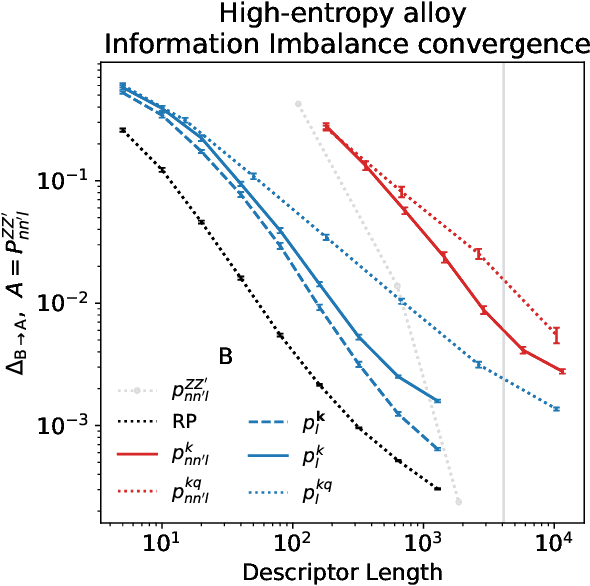
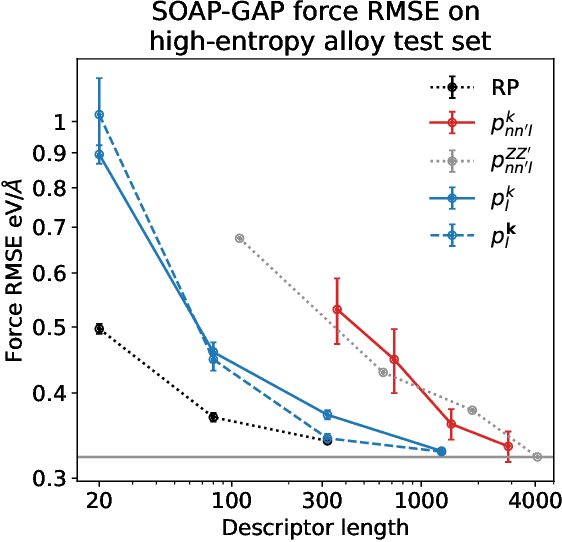
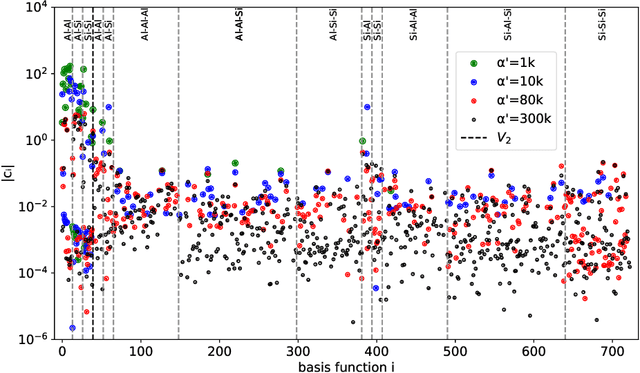
Density based representations of atomic environments that are invariant under Euclidean symmetries have become a widely used tool in the machine learning of interatomic potentials, broader data-driven atomistic modelling and the visualisation and analysis of materials datasets.The standard mechanism used to incorporate chemical element information is to create separate densities for each element and form tensor products between them. This leads to a steep scaling in the size of the representation as the number of elements increases. Graph neural networks, which do not explicitly use density representations, escape this scaling by mapping the chemical element information into a fixed dimensional space in a learnable way. We recast this approach as tensor factorisation by exploiting the tensor structure of standard neighbour density based descriptors. In doing so, we form compact tensor-reduced representations whose size does not depend on the number of chemical elements, but remain systematically convergeable and are therefore applicable to a wide range of data analysis and regression tasks.