 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCoupled reaction and diffusion governing interface evolution in solid-state batteries
Jun 12, 2025Understanding and controlling the atomistic-level reactions governing the formation of the solid-electrolyte interphase (SEI) is crucial for the viability of next-generation solid state batteries. However, challenges persist due to difficulties in experimentally characterizing buried interfaces and limits in simulation speed and accuracy. We conduct large-scale explicit reactive simulations with quantum accuracy for a symmetric battery cell, {\symcell}, enabled by active learning and deep equivariant neural network interatomic potentials. To automatically characterize the coupled reactions and interdiffusion at the interface, we formulate and use unsupervised classification techniques based on clustering in the space of local atomic environments. Our analysis reveals the formation of a previously unreported crystalline disordered phase, Li$_2$S$_{0.72}$P$_{0.14}$Cl$_{0.14}$, in the SEI, that evaded previous predictions based purely on thermodynamics, underscoring the importance of explicit modeling of full reaction and transport kinetics. Our simulations agree with and explain experimental observations of the SEI formations and elucidate the Li creep mechanisms, critical to dendrite initiation, characterized by significant Li motion along the interface. Our approach is to crease a digital twin from first principles, without adjustable parameters fitted to experiment. As such, it offers capabilities to gain insights into atomistic dynamics governing complex heterogeneous processes in solid-state synthesis and electrochemistry.
High-performance training and inference for deep equivariant interatomic potentials
Apr 22, 2025Machine learning interatomic potentials, particularly those based on deep equivariant neural networks, have demonstrated state-of-the-art accuracy and computational efficiency in atomistic modeling tasks like molecular dynamics and high-throughput screening. The size of datasets and demands of downstream workflows are growing rapidly, making robust and scalable software essential. This work presents a major overhaul of the NequIP framework focusing on multi-node parallelism, computational performance, and extensibility. The redesigned framework supports distributed training on large datasets and removes barriers preventing full utilization of the PyTorch 2.0 compiler at train time. We demonstrate this acceleration in a case study by training Allegro models on the SPICE 2 dataset of organic molecular systems. For inference, we introduce the first end-to-end infrastructure that uses the PyTorch Ahead-of-Time Inductor compiler for machine learning interatomic potentials. Additionally, we implement a custom kernel for the Allegro model's most expensive operation, the tensor product. Together, these advancements speed up molecular dynamics calculations on system sizes of practical relevance by up to a factor of 18.
A Recipe for Charge Density Prediction
May 29, 2024In density functional theory, charge density is the core attribute of atomic systems from which all chemical properties can be derived. Machine learning methods are promising in significantly accelerating charge density prediction, yet existing approaches either lack accuracy or scalability. We propose a recipe that can achieve both. In particular, we identify three key ingredients: (1) representing the charge density with atomic and virtual orbitals (spherical fields centered at atom/virtual coordinates); (2) using expressive and learnable orbital basis sets (basis function for the spherical fields); and (3) using high-capacity equivariant neural network architecture. Our method achieves state-of-the-art accuracy while being more than an order of magnitude faster than existing methods. Furthermore, our method enables flexible efficiency-accuracy trade-offs by adjusting the model/basis sizes.
Learning Interatomic Potentials at Multiple Scales
Oct 20, 2023The need to use a short time step is a key limit on the speed of molecular dynamics (MD) simulations. Simulations governed by classical potentials are often accelerated by using a multiple-time-step (MTS) integrator that evaluates certain potential energy terms that vary more slowly than others less frequently. This approach is enabled by the simple but limiting analytic forms of classical potentials. Machine learning interatomic potentials (MLIPs), in particular recent equivariant neural networks, are much more broadly applicable than classical potentials and can faithfully reproduce the expensive but accurate reference electronic structure calculations used to train them. They still, however, require the use of a single short time step, as they lack the inherent term-by-term scale separation of classical potentials. This work introduces a method to learn a scale separation in complex interatomic interactions by co-training two MLIPs. Initially, a small and efficient model is trained to reproduce short-time-scale interactions. Subsequently, a large and expressive model is trained jointly to capture the remaining interactions not captured by the small model. When running MD, the MTS integrator then evaluates the smaller model for every time step and the larger model less frequently, accelerating simulation. Compared to a conventionally trained MLIP, our approach can achieve a significant speedup (~3x in our experiments) without a loss of accuracy on the potential energy or simulation-derived quantities.
Scaling the leading accuracy of deep equivariant models to biomolecular simulations of realistic size
Apr 20, 2023
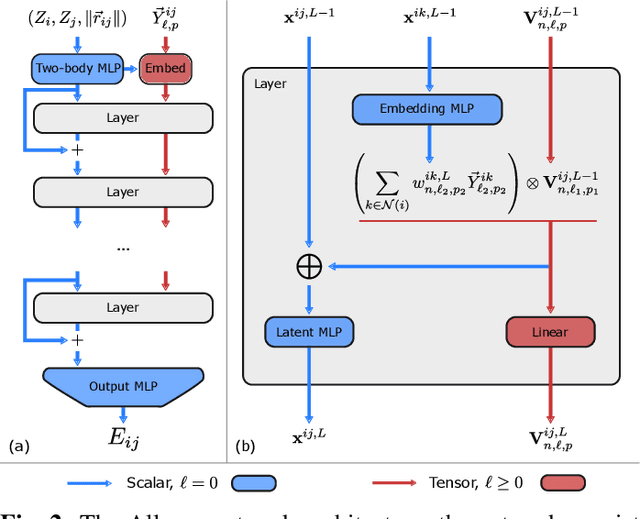
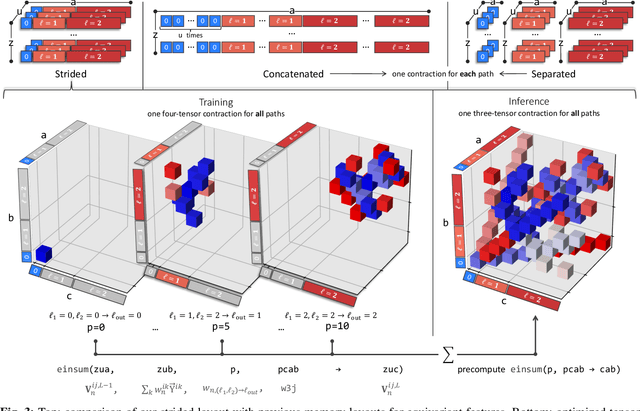
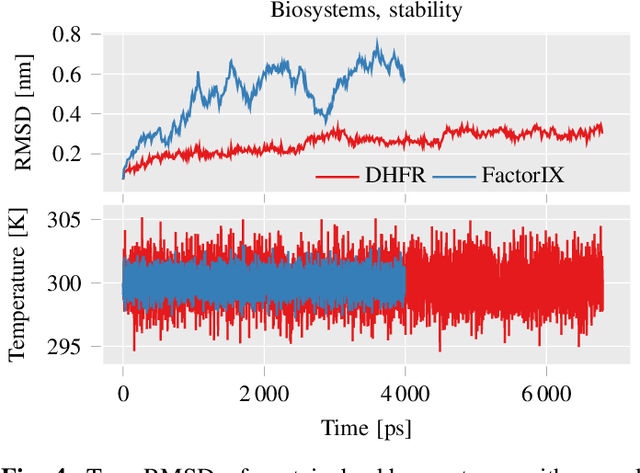
This work brings the leading accuracy, sample efficiency, and robustness of deep equivariant neural networks to the extreme computational scale. This is achieved through a combination of innovative model architecture, massive parallelization, and models and implementations optimized for efficient GPU utilization. The resulting Allegro architecture bridges the accuracy-speed tradeoff of atomistic simulations and enables description of dynamics in structures of unprecedented complexity at quantum fidelity. To illustrate the scalability of Allegro, we perform nanoseconds-long stable simulations of protein dynamics and scale up to a 44-million atom structure of a complete, all-atom, explicitly solvated HIV capsid on the Perlmutter supercomputer. We demonstrate excellent strong scaling up to 100 million atoms and 70% weak scaling to 5120 A100 GPUs.
Fast Uncertainty Estimates in Deep Learning Interatomic Potentials
Nov 17, 2022

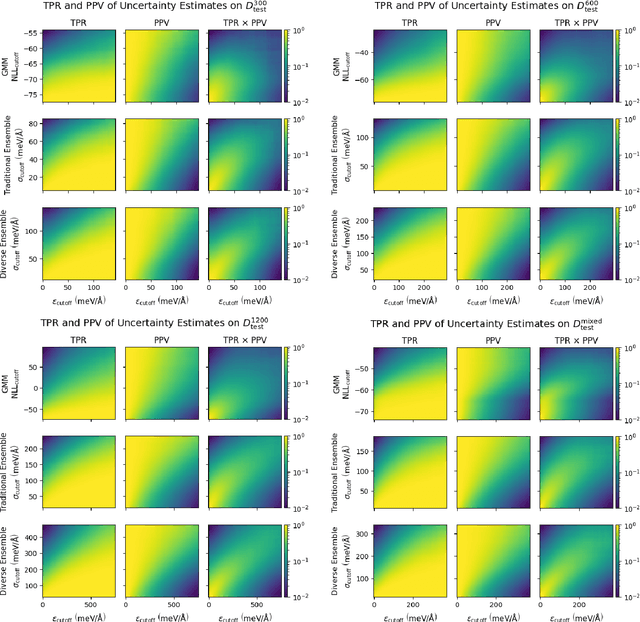

Deep learning has emerged as a promising paradigm to give access to highly accurate predictions of molecular and materials properties. A common short-coming shared by current approaches, however, is that neural networks only give point estimates of their predictions and do not come with predictive uncertainties associated with these estimates. Existing uncertainty quantification efforts have primarily leveraged the standard deviation of predictions across an ensemble of independently trained neural networks. This incurs a large computational overhead in both training and prediction that often results in order-of-magnitude more expensive predictions. Here, we propose a method to estimate the predictive uncertainty based on a single neural network without the need for an ensemble. This allows us to obtain uncertainty estimates with virtually no additional computational overhead over standard training and inference. We demonstrate that the quality of the uncertainty estimates matches those obtained from deep ensembles. We further examine the uncertainty estimates of our methods and deep ensembles across the configuration space of our test system and compare the uncertainties to the potential energy surface. Finally, we study the efficacy of the method in an active learning setting and find the results to match an ensemble-based strategy at order-of-magnitude reduced computational cost.
The Design Space of E-Equivariant Atom-Centered Interatomic Potentials
May 13, 2022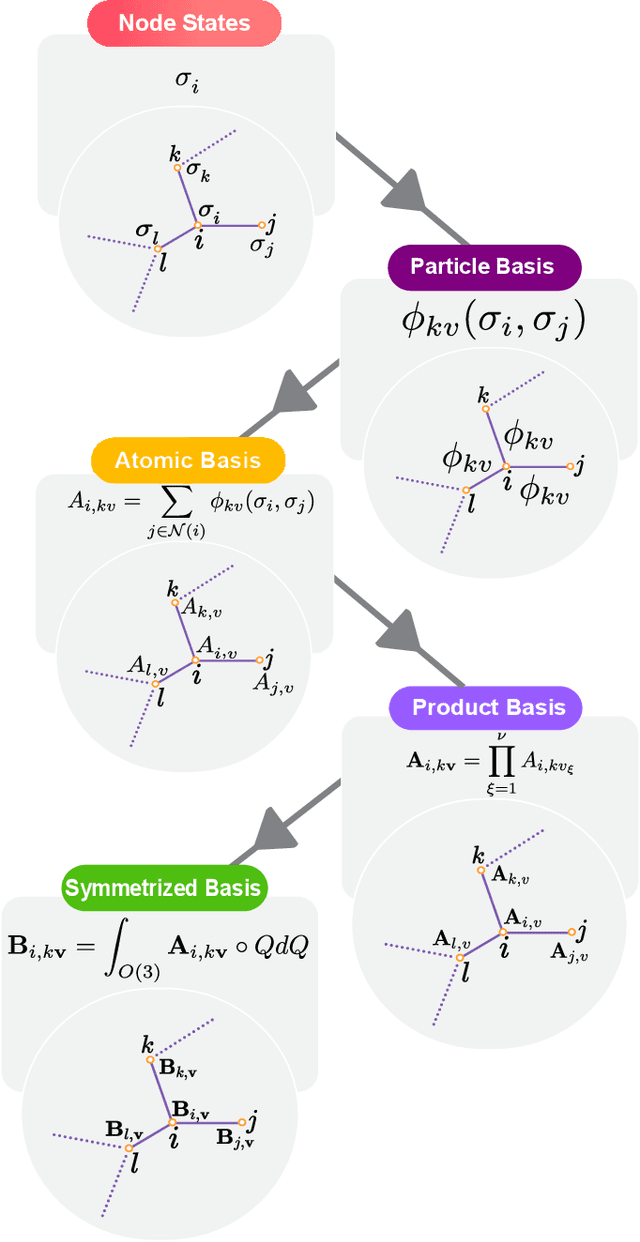
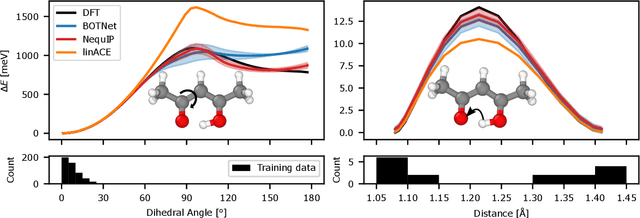
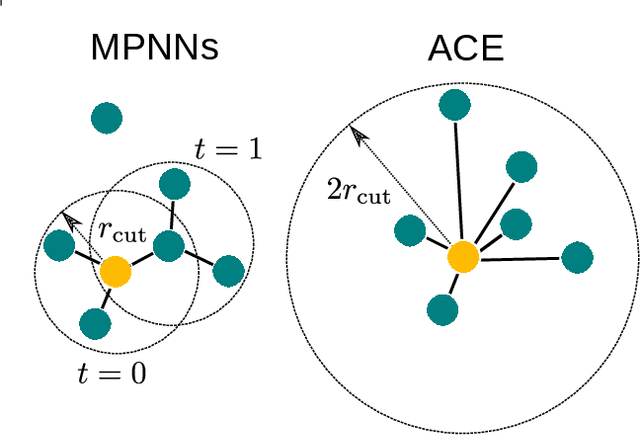
The rapid progress of machine learning interatomic potentials over the past couple of years produced a number of new architectures. Particularly notable among these are the Atomic Cluster Expansion (ACE), which unified many of the earlier ideas around atom density-based descriptors, and Neural Equivariant Interatomic Potentials (NequIP), a message passing neural network with equivariant features that showed state of the art accuracy. In this work, we construct a mathematical framework that unifies these models: ACE is generalised so that it can be recast as one layer of a multi-layer architecture. From another point of view, the linearised version of NequIP is understood as a particular sparsification of a much larger polynomial model. Our framework also provides a practical tool for systematically probing different choices in the unified design space. We demonstrate this by an ablation study of NequIP via a set of experiments looking at in- and out-of-domain accuracy and smooth extrapolation very far from the training data, and shed some light on which design choices are critical for achieving high accuracy. Finally, we present BOTNet (Body-Ordered-Tensor-Network), a much-simplified version of NequIP, which has an interpretable architecture and maintains accuracy on benchmark datasets.
Learning Local Equivariant Representations for Large-Scale Atomistic Dynamics
Apr 11, 2022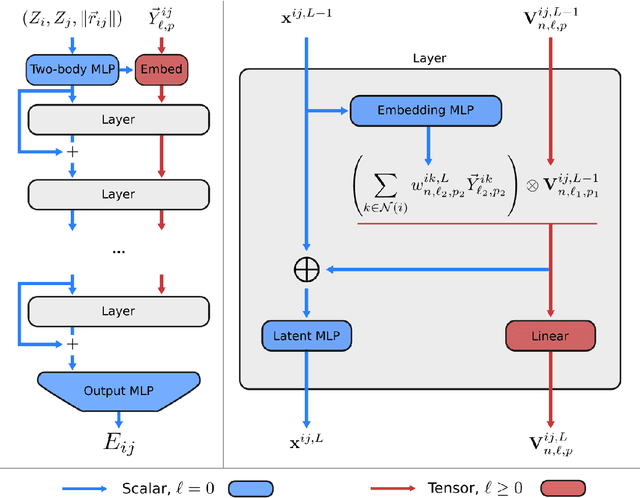
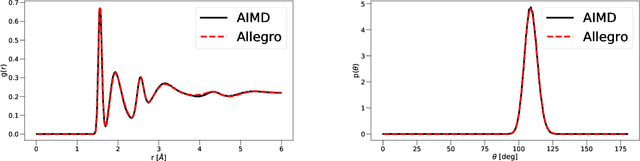
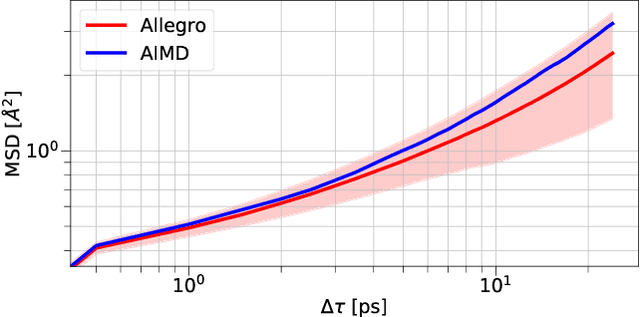
A simultaneously accurate and computationally efficient parametrization of the energy and atomic forces of molecules and materials is a long-standing goal in the natural sciences. In pursuit of this goal, neural message passing has lead to a paradigm shift by describing many-body correlations of atoms through iteratively passing messages along an atomistic graph. This propagation of information, however, makes parallel computation difficult and limits the length scales that can be studied. Strictly local descriptor-based methods, on the other hand, can scale to large systems but do not currently match the high accuracy observed with message passing approaches. This work introduces Allegro, a strictly local equivariant deep learning interatomic potential that simultaneously exhibits excellent accuracy and scalability of parallel computation. Allegro learns many-body functions of atomic coordinates using a series of tensor products of learned equivariant representations, but without relying on message passing. Allegro obtains improvements over state-of-the-art methods on the QM9 and revised MD-17 data sets. A single tensor product layer is shown to outperform existing deep message passing neural networks and transformers on the QM9 benchmark. Furthermore, Allegro displays remarkable generalization to out-of-distribution data. Molecular dynamics simulations based on Allegro recover structural and kinetic properties of an amorphous phosphate electrolyte in excellent agreement with first principles calculations. Finally, we demonstrate the parallel scaling of Allegro with a dynamics simulation of 100 million atoms.