 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnhanced Diffusion Sampling: Efficient Rare Event Sampling and Free Energy Calculation with Diffusion Models
Feb 18, 2026The rare-event sampling problem has long been the central limiting factor in molecular dynamics (MD), especially in biomolecular simulation. Recently, diffusion models such as BioEmu have emerged as powerful equilibrium samplers that generate independent samples from complex molecular distributions, eliminating the cost of sampling rare transition events. However, a sampling problem remains when computing observables that rely on states which are rare in equilibrium, for example folding free energies. Here, we introduce enhanced diffusion sampling, enabling efficient exploration of rare-event regions while preserving unbiased thermodynamic estimators. The key idea is to perform quantitatively accurate steering protocols to generate biased ensembles and subsequently recover equilibrium statistics via exact reweighting. We instantiate our framework in three algorithms: UmbrellaDiff (umbrella sampling with diffusion models), $Δ$G-Diff (free-energy differences via tilted ensembles), and MetaDiff (a batchwise analogue for metadynamics). Across toy systems, protein folding landscapes and folding free energies, our methods achieve fast, accurate, and scalable estimation of equilibrium properties within GPU-minutes to hours per system -- closing the rare-event sampling gap that remained after the advent of diffusion-model equilibrium samplers.
MatterGen: a generative model for inorganic materials design
Dec 06, 2023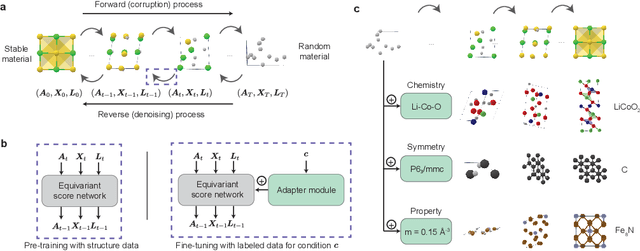
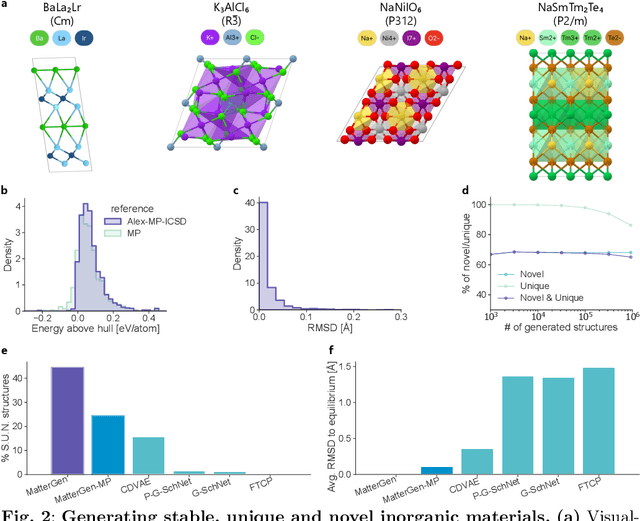
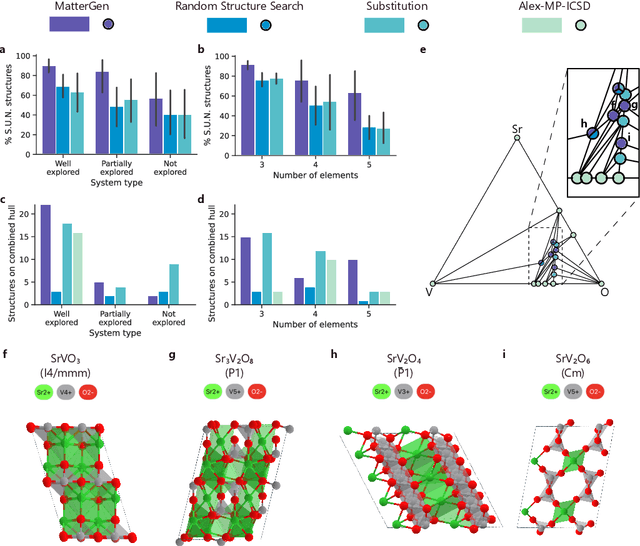
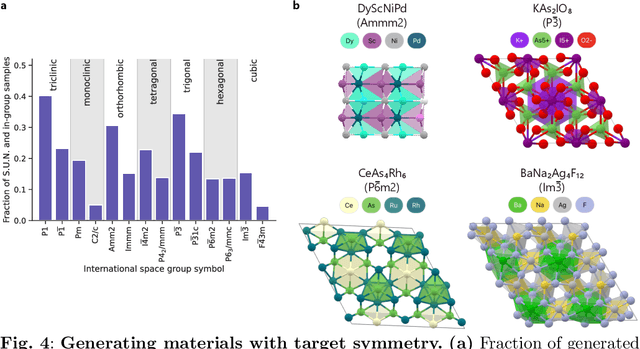
The design of functional materials with desired properties is essential in driving technological advances in areas like energy storage, catalysis, and carbon capture. Generative models provide a new paradigm for materials design by directly generating entirely novel materials given desired property constraints. Despite recent progress, current generative models have low success rate in proposing stable crystals, or can only satisfy a very limited set of property constraints. Here, we present MatterGen, a model that generates stable, diverse inorganic materials across the periodic table and can further be fine-tuned to steer the generation towards a broad range of property constraints. To enable this, we introduce a new diffusion-based generative process that produces crystalline structures by gradually refining atom types, coordinates, and the periodic lattice. We further introduce adapter modules to enable fine-tuning towards any given property constraints with a labeled dataset. Compared to prior generative models, structures produced by MatterGen are more than twice as likely to be novel and stable, and more than 15 times closer to the local energy minimum. After fine-tuning, MatterGen successfully generates stable, novel materials with desired chemistry, symmetry, as well as mechanical, electronic and magnetic properties. Finally, we demonstrate multi-property materials design capabilities by proposing structures that have both high magnetic density and a chemical composition with low supply-chain risk. We believe that the quality of generated materials and the breadth of MatterGen's capabilities represent a major advancement towards creating a universal generative model for materials design.
Does AI for science need another ImageNet Or totally different benchmarks? A case study of machine learning force fields
Aug 11, 2023



AI for science (AI4S) is an emerging research field that aims to enhance the accuracy and speed of scientific computing tasks using machine learning methods. Traditional AI benchmarking methods struggle to adapt to the unique challenges posed by AI4S because they assume data in training, testing, and future real-world queries are independent and identically distributed, while AI4S workloads anticipate out-of-distribution problem instances. This paper investigates the need for a novel approach to effectively benchmark AI for science, using the machine learning force field (MLFF) as a case study. MLFF is a method to accelerate molecular dynamics (MD) simulation with low computational cost and high accuracy. We identify various missed opportunities in scientifically meaningful benchmarking and propose solutions to evaluate MLFF models, specifically in the aspects of sample efficiency, time domain sensitivity, and cross-dataset generalization capabilities. By setting up the problem instantiation similar to the actual scientific applications, more meaningful performance metrics from the benchmark can be achieved. This suite of metrics has demonstrated a better ability to assess a model's performance in real-world scientific applications, in contrast to traditional AI benchmarking methodologies. This work is a component of the SAIBench project, an AI4S benchmarking suite. The project homepage is https://www.computercouncil.org/SAIBench.
Learning Local Equivariant Representations for Large-Scale Atomistic Dynamics
Apr 11, 2022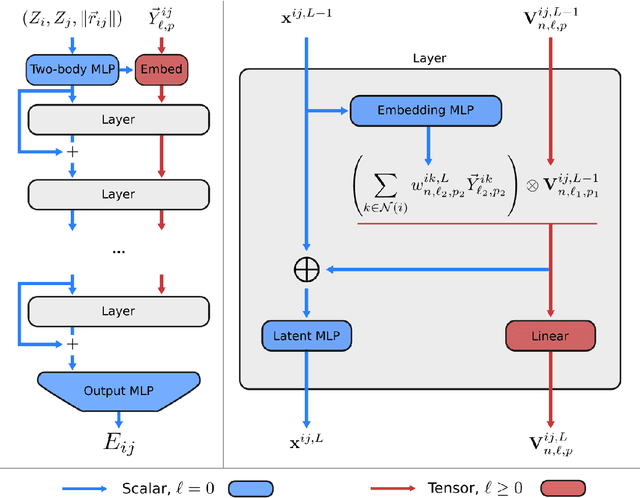
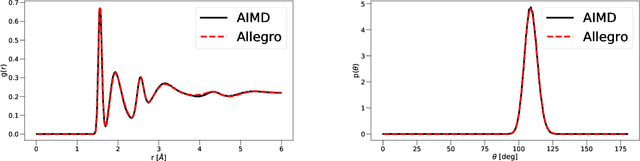
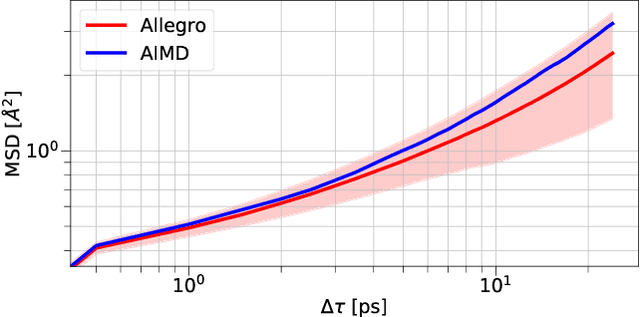
A simultaneously accurate and computationally efficient parametrization of the energy and atomic forces of molecules and materials is a long-standing goal in the natural sciences. In pursuit of this goal, neural message passing has lead to a paradigm shift by describing many-body correlations of atoms through iteratively passing messages along an atomistic graph. This propagation of information, however, makes parallel computation difficult and limits the length scales that can be studied. Strictly local descriptor-based methods, on the other hand, can scale to large systems but do not currently match the high accuracy observed with message passing approaches. This work introduces Allegro, a strictly local equivariant deep learning interatomic potential that simultaneously exhibits excellent accuracy and scalability of parallel computation. Allegro learns many-body functions of atomic coordinates using a series of tensor products of learned equivariant representations, but without relying on message passing. Allegro obtains improvements over state-of-the-art methods on the QM9 and revised MD-17 data sets. A single tensor product layer is shown to outperform existing deep message passing neural networks and transformers on the QM9 benchmark. Furthermore, Allegro displays remarkable generalization to out-of-distribution data. Molecular dynamics simulations based on Allegro recover structural and kinetic properties of an amorphous phosphate electrolyte in excellent agreement with first principles calculations. Finally, we demonstrate the parallel scaling of Allegro with a dynamics simulation of 100 million atoms.
SE-Equivariant Graph Neural Networks for Data-Efficient and Accurate Interatomic Potentials
Jan 08, 2021
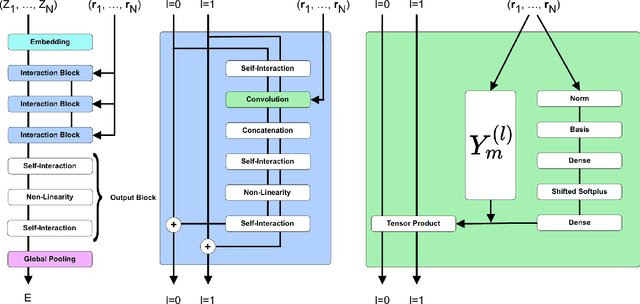
This work presents Neural Equivariant Interatomic Potentials (NequIP), a SE(3)-equivariant neural network approach for learning interatomic potentials from ab-initio calculations for molecular dynamics simulations. While most contemporary symmetry-aware models use invariant convolutions and only act on scalars, NequIP employs SE(3)-equivariant convolutions for interactions of geometric tensors, resulting in a more information-rich and faithful representation of atomic environments. The method achieves state-of-the-art accuracy on a challenging set of diverse molecules and materials while exhibiting remarkable data efficiency. NequIP outperforms existing models with up to three orders of magnitude fewer training data, challenging the widely held belief that deep neural networks require massive training sets. The high data efficiency of the method allows for the construction of accurate potentials using high-order quantum chemical level of theory as reference and enables high-fidelity molecular dynamics simulations over long time scales.
Multitask machine learning of collective variables for enhanced sampling of rare events
Dec 07, 2020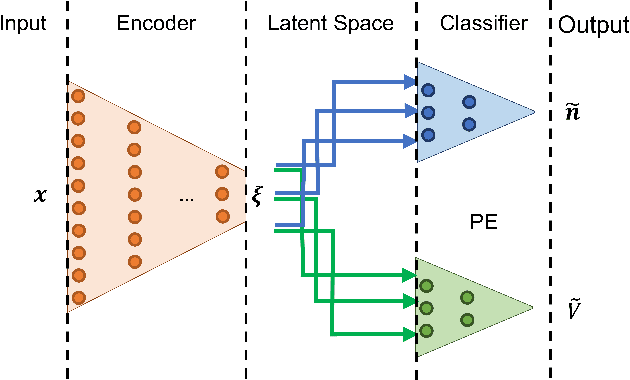
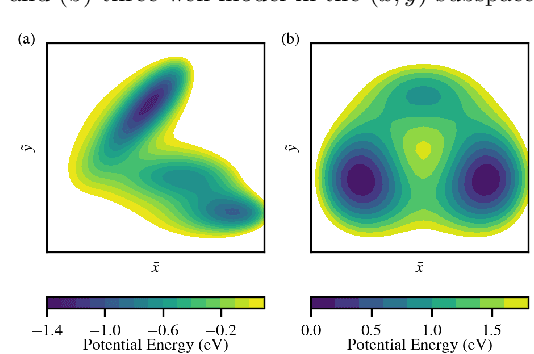
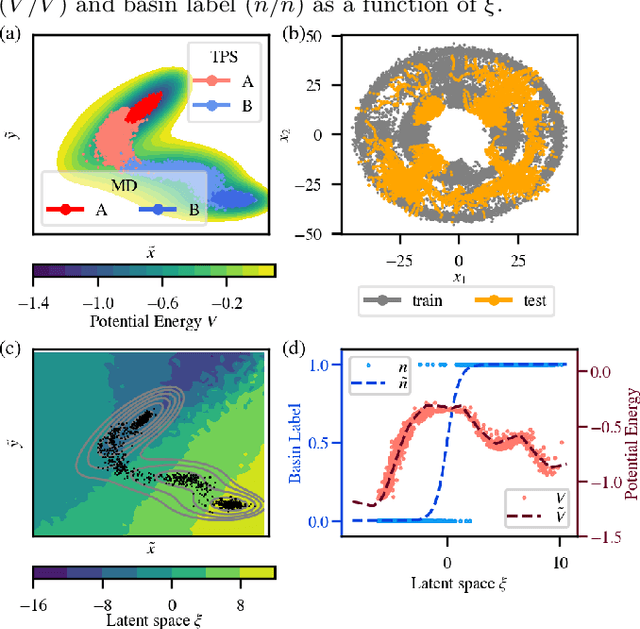
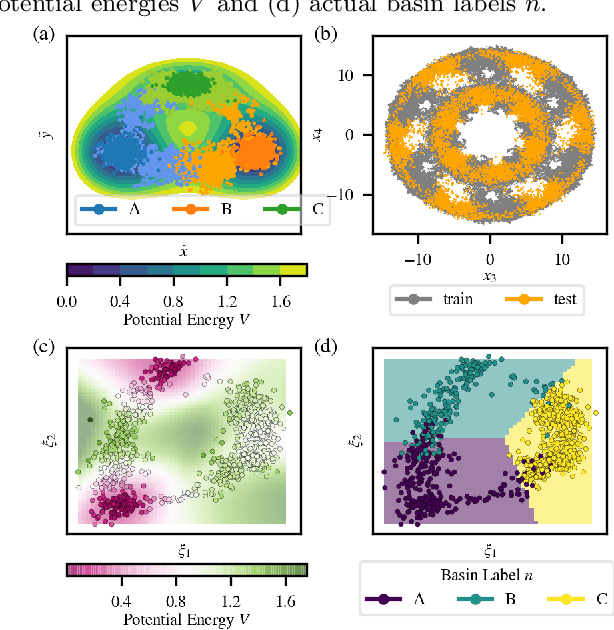
Computing accurate reaction rates is a central challenge in computational chemistry and biology because of the high cost of free energy estimation with unbiased molecular dynamics. In this work, a data-driven machine learning algorithm is devised to learn collective variables with a multitask neural network, where a common upstream part reduces the high dimensionality of atomic configurations to a low dimensional latent space, and separate downstream parts map the latent space to predictions of basin class labels and potential energies. The resulting latent space is shown to be an effective low-dimensional representation, capturing the reaction progress and guiding effective umbrella sampling to obtain accurate free energy landscapes. This approach is successfully applied to model systems including a 5D M\"uller Brown model, a 5D three-well model, and alanine dipeptide in vacuum. This approach enables automated dimensionality reduction for energy controlled reactions in complex systems, offers a unified framework that can be trained with limited data, and outperforms single-task learning approaches, including autoencoders.
Fast Bayesian Force Fields from Active Learning: Study of Inter-Dimensional Transformation of Stanene
Aug 26, 2020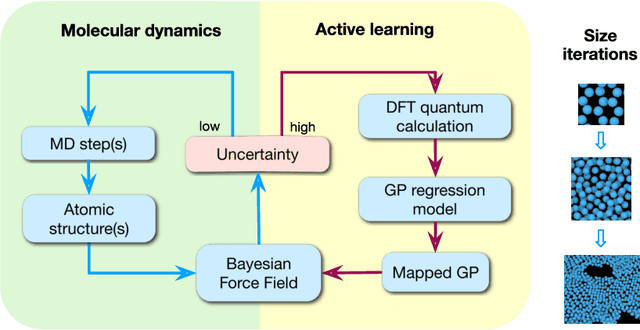
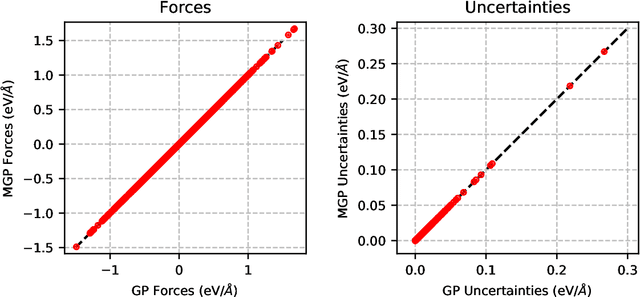
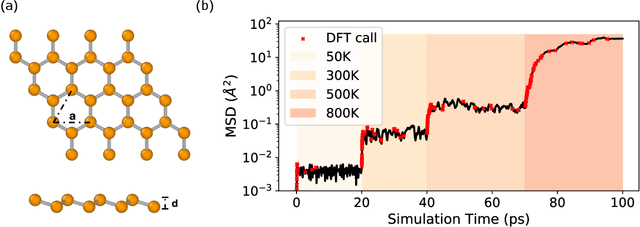
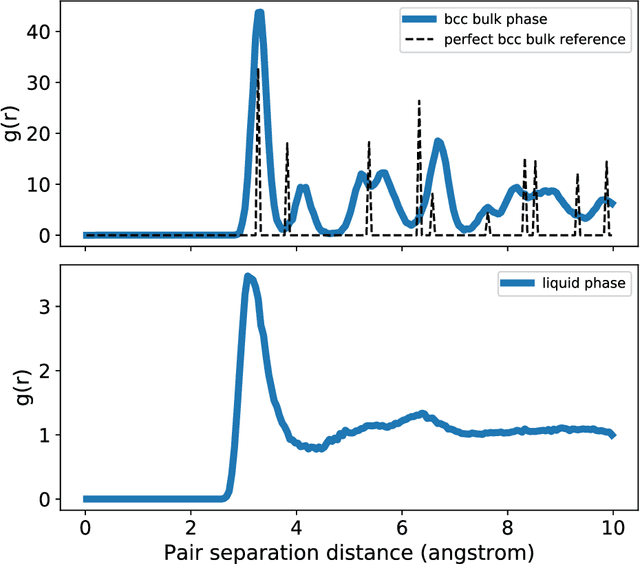
We present a way to dramatically accelerate Gaussian process models for interatomic force fields based on many-body kernels by mapping both forces and uncertainties onto functions of low-dimensional features. This allows for automated active learning of models combining near-quantum accuracy, built-in uncertainty, and constant cost of evaluation that is comparable to classical analytical models, capable of simulating millions of atoms. Using this approach, we perform large scale molecular dynamics simulations of the stability of the stanene monolayer. We discover an unusual phase transformation mechanism of 2D stanene, where ripples lead to nucleation of bilayer defects, densification into a disordered multilayer structure, followed by formation of bulk liquid at high temperature or nucleation and growth of the 3D bcc crystal at low temperature. The presented method opens possibilities for rapid development of fast accurate uncertainty-aware models for simulating long-time large-scale dynamics of complex materials.