 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSE-Equivariant Graph Neural Networks for Data-Efficient and Accurate Interatomic Potentials
Jan 08, 2021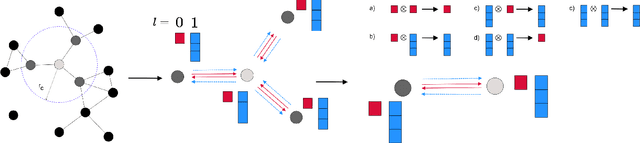
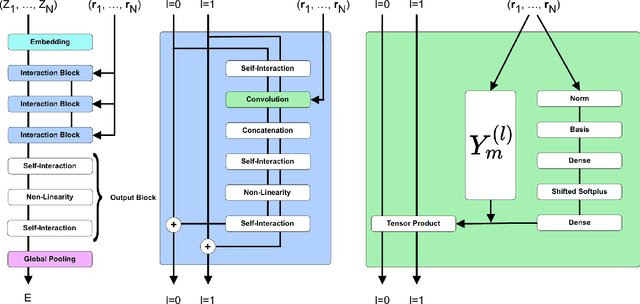
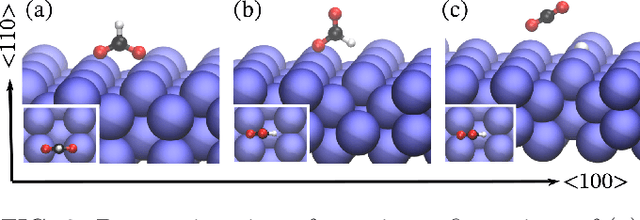
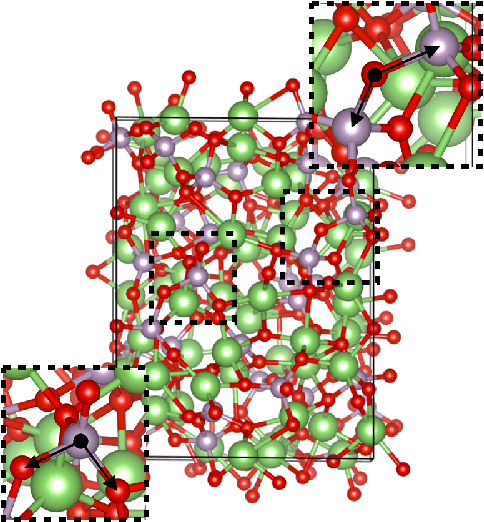
This work presents Neural Equivariant Interatomic Potentials (NequIP), a SE(3)-equivariant neural network approach for learning interatomic potentials from ab-initio calculations for molecular dynamics simulations. While most contemporary symmetry-aware models use invariant convolutions and only act on scalars, NequIP employs SE(3)-equivariant convolutions for interactions of geometric tensors, resulting in a more information-rich and faithful representation of atomic environments. The method achieves state-of-the-art accuracy on a challenging set of diverse molecules and materials while exhibiting remarkable data efficiency. NequIP outperforms existing models with up to three orders of magnitude fewer training data, challenging the widely held belief that deep neural networks require massive training sets. The high data efficiency of the method allows for the construction of accurate potentials using high-order quantum chemical level of theory as reference and enables high-fidelity molecular dynamics simulations over long time scales.
Fast Neural Network Approach for Direct Covariant Forces Prediction in Complex Multi-Element Extended Systems
May 07, 2019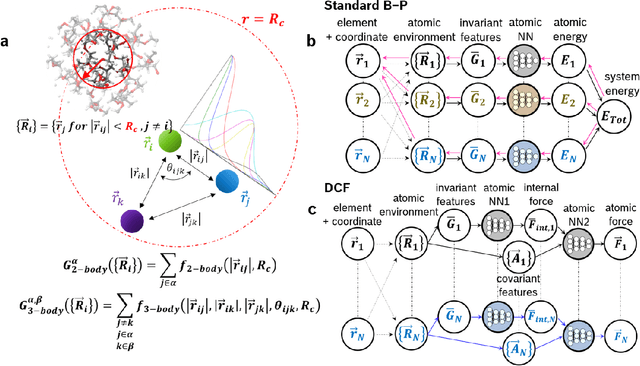
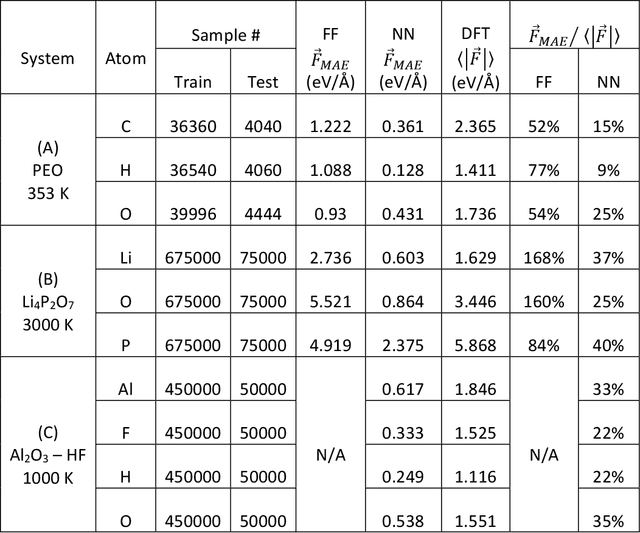
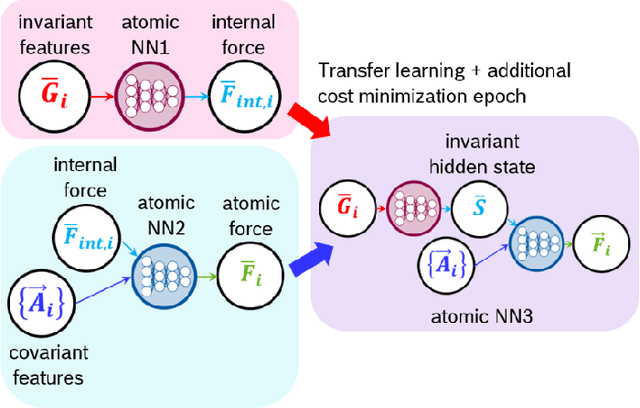
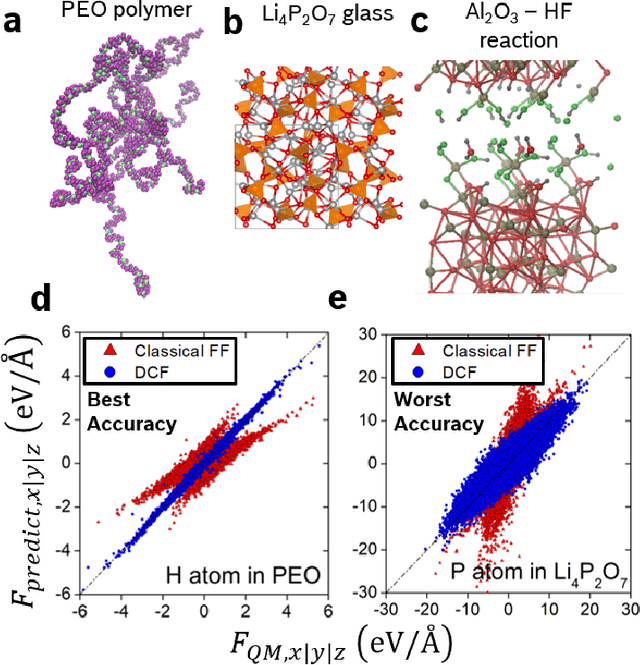
Neural network force field (NNFF) is a method for performing regression on atomic structure-force relationships, bypassing expensive quantum mechanics calculation which prevents the execution of long ab-initio quality molecular dynamics simulations. However, most NNFF methods for complex multi-element atomic systems indirectly predict atomic force vectors by exploiting just atomic structure rotation-invariant features and the network-feature spatial derivatives which are computationally expensive. We develop a staggered NNFF architecture exploiting both rotation-invariant and covariant features separately to directly predict atomic force vectors without using spatial derivatives, thereby reducing expensive structural feature calculation by ~180-480x. This acceleration enables us to develop NNFF which directly predicts atomic forces in complex ternary and quaternary-element extended systems comprised of long polymer chains, amorphous oxide, and surface chemical reactions. The staggered rotation-invariant-covariant architecture described here can also directly predict complex covariant vector outputs from local physical structures in domains beyond computational material science.