 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFast Neural Network Approach for Direct Covariant Forces Prediction in Complex Multi-Element Extended Systems
May 07, 2019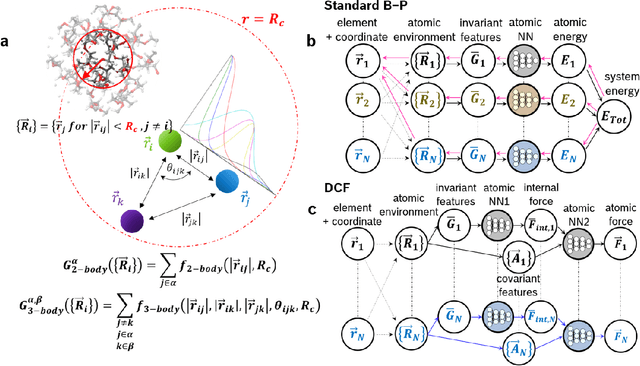
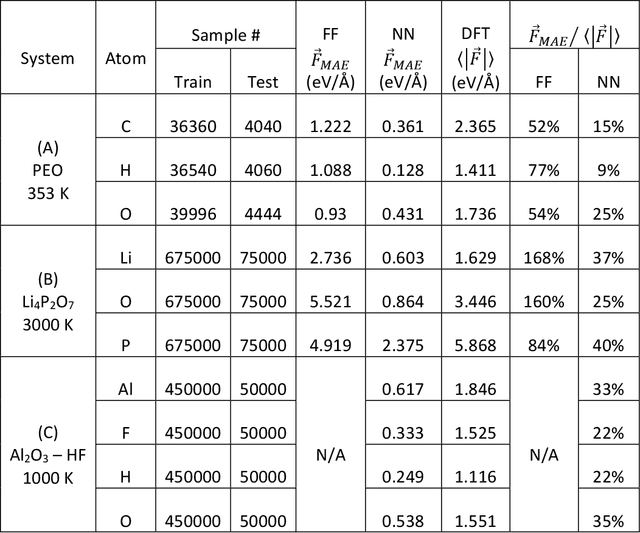
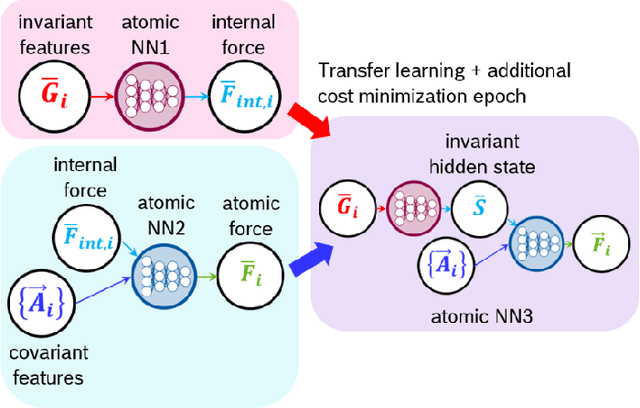
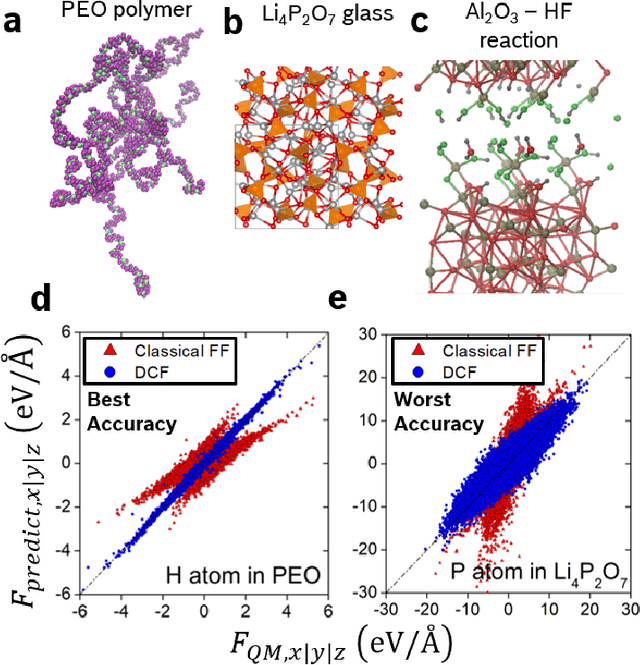
Neural network force field (NNFF) is a method for performing regression on atomic structure-force relationships, bypassing expensive quantum mechanics calculation which prevents the execution of long ab-initio quality molecular dynamics simulations. However, most NNFF methods for complex multi-element atomic systems indirectly predict atomic force vectors by exploiting just atomic structure rotation-invariant features and the network-feature spatial derivatives which are computationally expensive. We develop a staggered NNFF architecture exploiting both rotation-invariant and covariant features separately to directly predict atomic force vectors without using spatial derivatives, thereby reducing expensive structural feature calculation by ~180-480x. This acceleration enables us to develop NNFF which directly predicts atomic forces in complex ternary and quaternary-element extended systems comprised of long polymer chains, amorphous oxide, and surface chemical reactions. The staggered rotation-invariant-covariant architecture described here can also directly predict complex covariant vector outputs from local physical structures in domains beyond computational material science.