 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGenerative Hierarchical Materials Search
Sep 10, 2024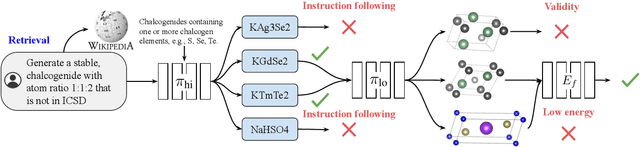


Generative models trained at scale can now produce text, video, and more recently, scientific data such as crystal structures. In applications of generative approaches to materials science, and in particular to crystal structures, the guidance from the domain expert in the form of high-level instructions can be essential for an automated system to output candidate crystals that are viable for downstream research. In this work, we formulate end-to-end language-to-structure generation as a multi-objective optimization problem, and propose Generative Hierarchical Materials Search (GenMS) for controllable generation of crystal structures. GenMS consists of (1) a language model that takes high-level natural language as input and generates intermediate textual information about a crystal (e.g., chemical formulae), and (2) a diffusion model that takes intermediate information as input and generates low-level continuous value crystal structures. GenMS additionally uses a graph neural network to predict properties (e.g., formation energy) from the generated crystal structures. During inference, GenMS leverages all three components to conduct a forward tree search over the space of possible structures. Experiments show that GenMS outperforms other alternatives of directly using language models to generate structures both in satisfying user request and in generating low-energy structures. We confirm that GenMS is able to generate common crystal structures such as double perovskites, or spinels, solely from natural language input, and hence can form the foundation for more complex structure generation in near future.
Predicting emergence of crystals from amorphous matter with deep learning
Oct 02, 2023Crystallization of the amorphous phases into metastable crystals plays a fundamental role in the formation of new matter, from geological to biological processes in nature to synthesis and development of new materials in the laboratory. Predicting the outcome of such phase transitions reliably would enable new research directions in these areas, but has remained beyond reach with molecular modeling or ab-initio methods. Here, we show that crystallization products of amorphous phases can be predicted in any inorganic chemistry by sampling the crystallization pathways of their local structural motifs at the atomistic level using universal deep learning potentials. We show that this approach identifies the crystal structures of polymorphs that initially nucleate from amorphous precursors with high accuracy across a diverse set of material systems, including polymorphic oxides, nitrides, carbides, fluorides, chlorides, chalcogenides, and metal alloys. Our results demonstrate that Ostwald's rule of stages can be exploited mechanistically at the molecular level to predictably access new metastable crystals from the amorphous phase in material synthesis.
Scaling the leading accuracy of deep equivariant models to biomolecular simulations of realistic size
Apr 20, 2023
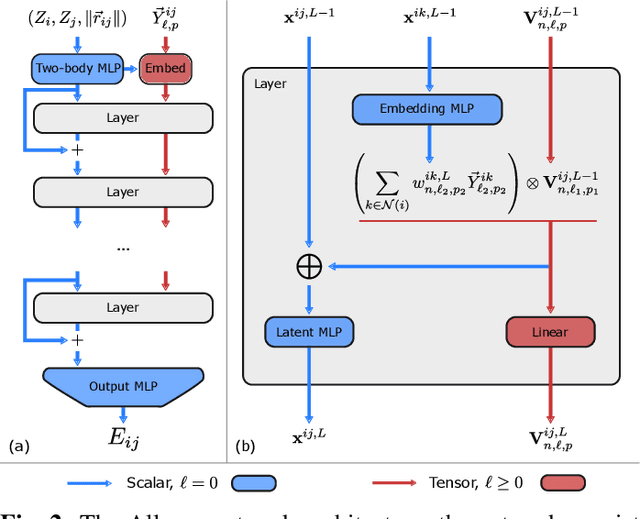
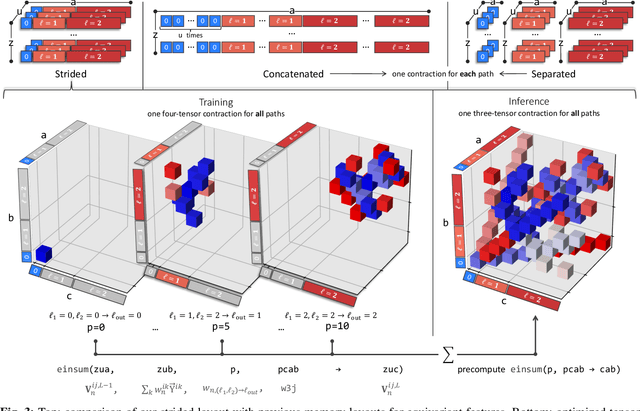
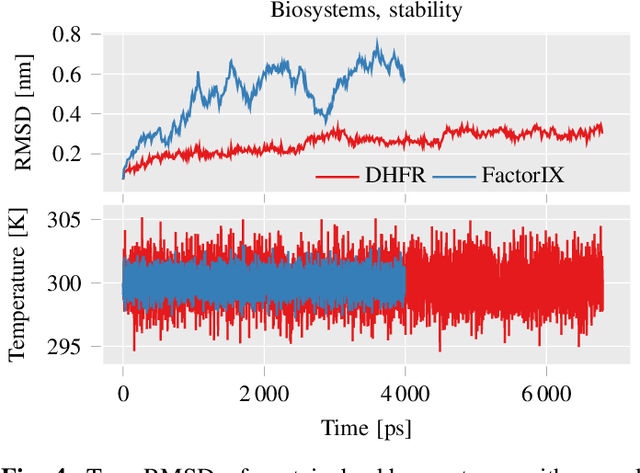
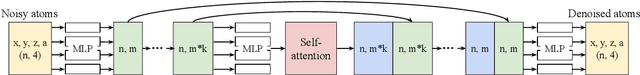
This work brings the leading accuracy, sample efficiency, and robustness of deep equivariant neural networks to the extreme computational scale. This is achieved through a combination of innovative model architecture, massive parallelization, and models and implementations optimized for efficient GPU utilization. The resulting Allegro architecture bridges the accuracy-speed tradeoff of atomistic simulations and enables description of dynamics in structures of unprecedented complexity at quantum fidelity. To illustrate the scalability of Allegro, we perform nanoseconds-long stable simulations of protein dynamics and scale up to a 44-million atom structure of a complete, all-atom, explicitly solvated HIV capsid on the Perlmutter supercomputer. We demonstrate excellent strong scaling up to 100 million atoms and 70% weak scaling to 5120 A100 GPUs.
Fast Uncertainty Estimates in Deep Learning Interatomic Potentials
Nov 17, 2022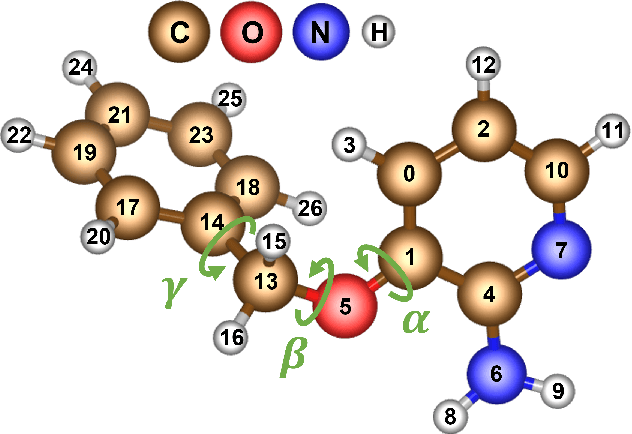

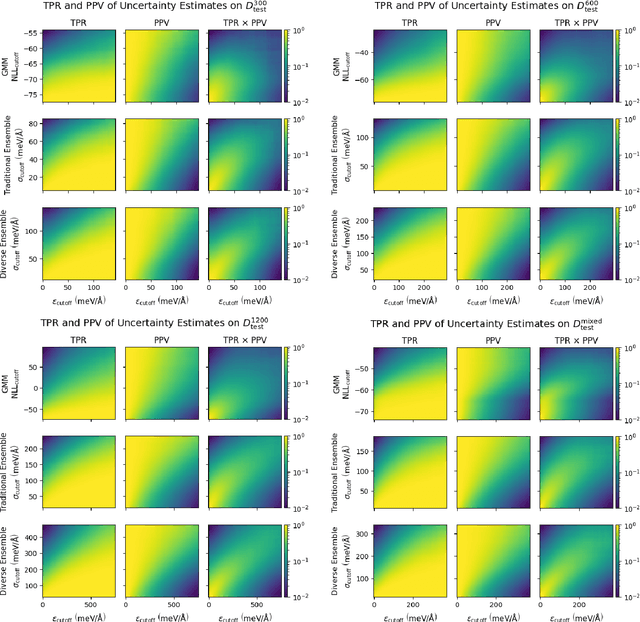

Deep learning has emerged as a promising paradigm to give access to highly accurate predictions of molecular and materials properties. A common short-coming shared by current approaches, however, is that neural networks only give point estimates of their predictions and do not come with predictive uncertainties associated with these estimates. Existing uncertainty quantification efforts have primarily leveraged the standard deviation of predictions across an ensemble of independently trained neural networks. This incurs a large computational overhead in both training and prediction that often results in order-of-magnitude more expensive predictions. Here, we propose a method to estimate the predictive uncertainty based on a single neural network without the need for an ensemble. This allows us to obtain uncertainty estimates with virtually no additional computational overhead over standard training and inference. We demonstrate that the quality of the uncertainty estimates matches those obtained from deep ensembles. We further examine the uncertainty estimates of our methods and deep ensembles across the configuration space of our test system and compare the uncertainties to the potential energy surface. Finally, we study the efficacy of the method in an active learning setting and find the results to match an ensemble-based strategy at order-of-magnitude reduced computational cost.
The Design Space of E-Equivariant Atom-Centered Interatomic Potentials
May 13, 2022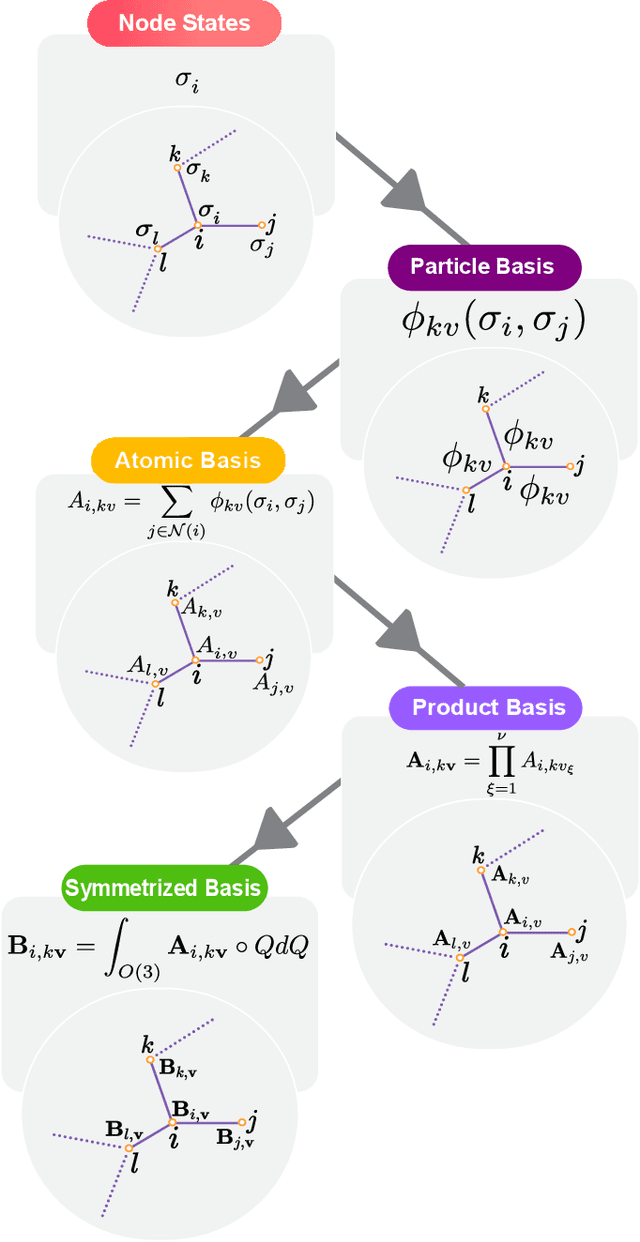
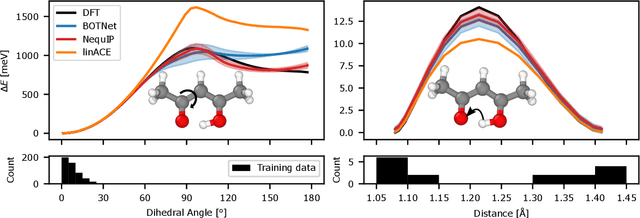
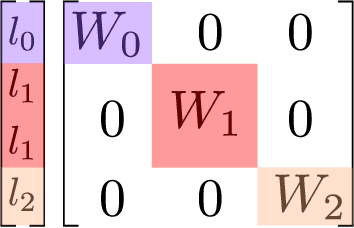
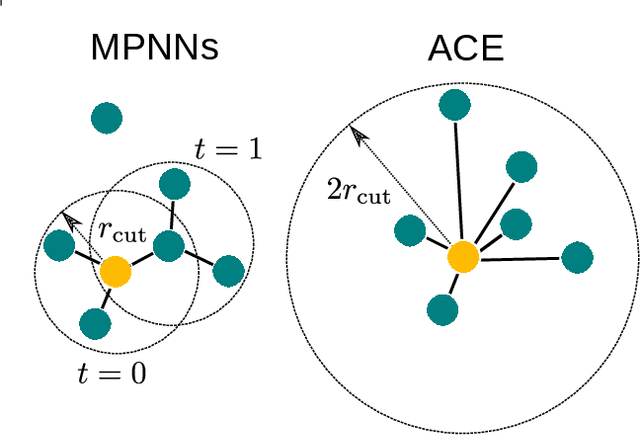
The rapid progress of machine learning interatomic potentials over the past couple of years produced a number of new architectures. Particularly notable among these are the Atomic Cluster Expansion (ACE), which unified many of the earlier ideas around atom density-based descriptors, and Neural Equivariant Interatomic Potentials (NequIP), a message passing neural network with equivariant features that showed state of the art accuracy. In this work, we construct a mathematical framework that unifies these models: ACE is generalised so that it can be recast as one layer of a multi-layer architecture. From another point of view, the linearised version of NequIP is understood as a particular sparsification of a much larger polynomial model. Our framework also provides a practical tool for systematically probing different choices in the unified design space. We demonstrate this by an ablation study of NequIP via a set of experiments looking at in- and out-of-domain accuracy and smooth extrapolation very far from the training data, and shed some light on which design choices are critical for achieving high accuracy. Finally, we present BOTNet (Body-Ordered-Tensor-Network), a much-simplified version of NequIP, which has an interpretable architecture and maintains accuracy on benchmark datasets.
Learning Local Equivariant Representations for Large-Scale Atomistic Dynamics
Apr 11, 2022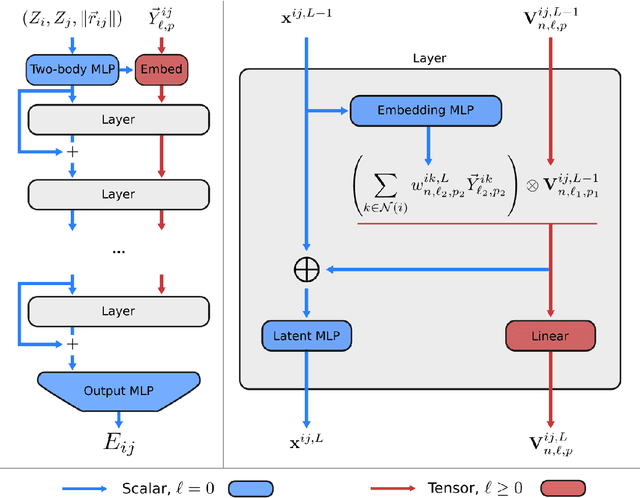
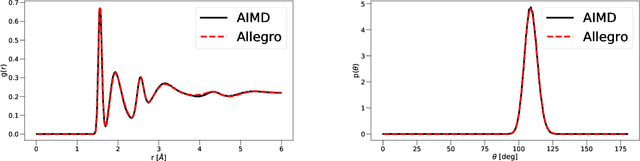
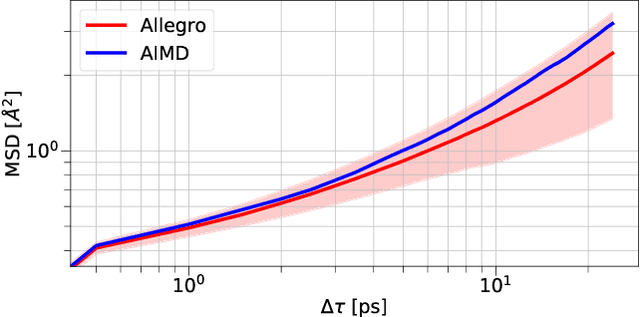
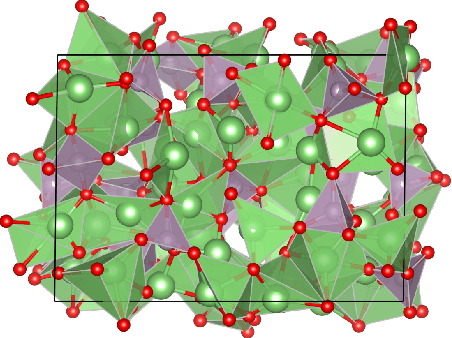
A simultaneously accurate and computationally efficient parametrization of the energy and atomic forces of molecules and materials is a long-standing goal in the natural sciences. In pursuit of this goal, neural message passing has lead to a paradigm shift by describing many-body correlations of atoms through iteratively passing messages along an atomistic graph. This propagation of information, however, makes parallel computation difficult and limits the length scales that can be studied. Strictly local descriptor-based methods, on the other hand, can scale to large systems but do not currently match the high accuracy observed with message passing approaches. This work introduces Allegro, a strictly local equivariant deep learning interatomic potential that simultaneously exhibits excellent accuracy and scalability of parallel computation. Allegro learns many-body functions of atomic coordinates using a series of tensor products of learned equivariant representations, but without relying on message passing. Allegro obtains improvements over state-of-the-art methods on the QM9 and revised MD-17 data sets. A single tensor product layer is shown to outperform existing deep message passing neural networks and transformers on the QM9 benchmark. Furthermore, Allegro displays remarkable generalization to out-of-distribution data. Molecular dynamics simulations based on Allegro recover structural and kinetic properties of an amorphous phosphate electrolyte in excellent agreement with first principles calculations. Finally, we demonstrate the parallel scaling of Allegro with a dynamics simulation of 100 million atoms.
SE-Equivariant Graph Neural Networks for Data-Efficient and Accurate Interatomic Potentials
Jan 08, 2021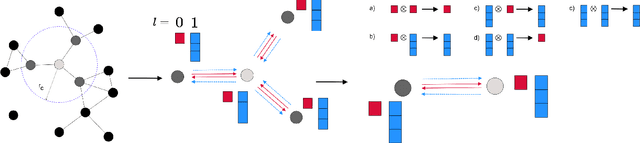
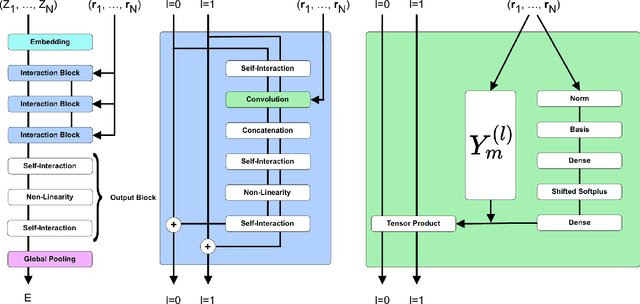
This work presents Neural Equivariant Interatomic Potentials (NequIP), a SE(3)-equivariant neural network approach for learning interatomic potentials from ab-initio calculations for molecular dynamics simulations. While most contemporary symmetry-aware models use invariant convolutions and only act on scalars, NequIP employs SE(3)-equivariant convolutions for interactions of geometric tensors, resulting in a more information-rich and faithful representation of atomic environments. The method achieves state-of-the-art accuracy on a challenging set of diverse molecules and materials while exhibiting remarkable data efficiency. NequIP outperforms existing models with up to three orders of magnitude fewer training data, challenging the widely held belief that deep neural networks require massive training sets. The high data efficiency of the method allows for the construction of accurate potentials using high-order quantum chemical level of theory as reference and enables high-fidelity molecular dynamics simulations over long time scales.
Multitask machine learning of collective variables for enhanced sampling of rare events
Dec 07, 2020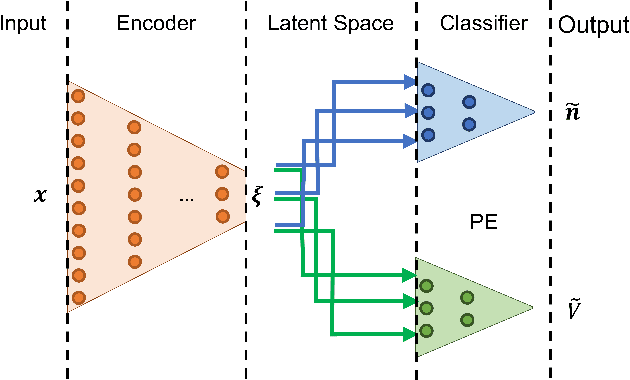
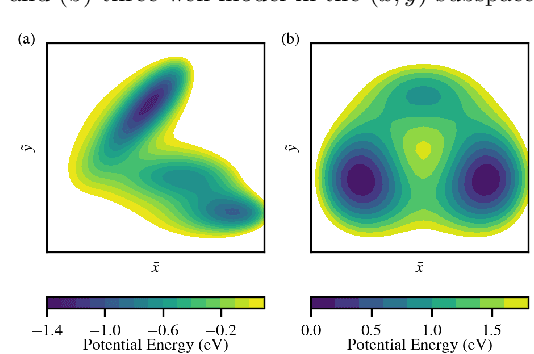
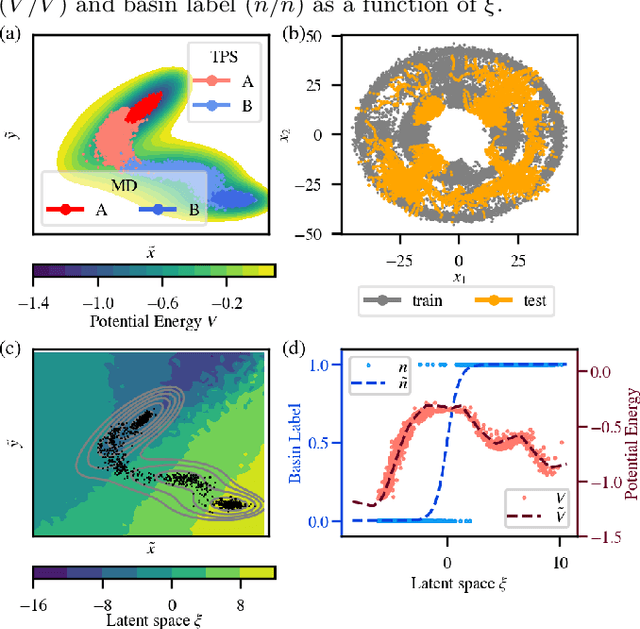
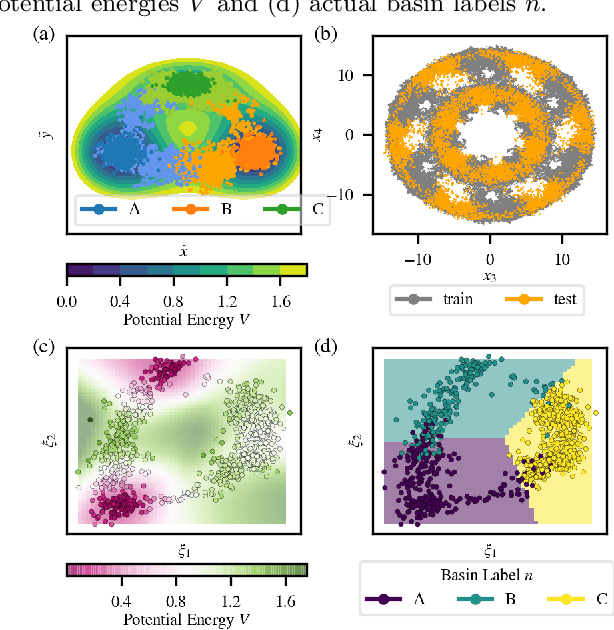
Computing accurate reaction rates is a central challenge in computational chemistry and biology because of the high cost of free energy estimation with unbiased molecular dynamics. In this work, a data-driven machine learning algorithm is devised to learn collective variables with a multitask neural network, where a common upstream part reduces the high dimensionality of atomic configurations to a low dimensional latent space, and separate downstream parts map the latent space to predictions of basin class labels and potential energies. The resulting latent space is shown to be an effective low-dimensional representation, capturing the reaction progress and guiding effective umbrella sampling to obtain accurate free energy landscapes. This approach is successfully applied to model systems including a 5D M\"uller Brown model, a 5D three-well model, and alanine dipeptide in vacuum. This approach enables automated dimensionality reduction for energy controlled reactions in complex systems, offers a unified framework that can be trained with limited data, and outperforms single-task learning approaches, including autoencoders.