 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePredicting emergence of crystals from amorphous matter with deep learning
Oct 02, 2023Crystallization of the amorphous phases into metastable crystals plays a fundamental role in the formation of new matter, from geological to biological processes in nature to synthesis and development of new materials in the laboratory. Predicting the outcome of such phase transitions reliably would enable new research directions in these areas, but has remained beyond reach with molecular modeling or ab-initio methods. Here, we show that crystallization products of amorphous phases can be predicted in any inorganic chemistry by sampling the crystallization pathways of their local structural motifs at the atomistic level using universal deep learning potentials. We show that this approach identifies the crystal structures of polymorphs that initially nucleate from amorphous precursors with high accuracy across a diverse set of material systems, including polymorphic oxides, nitrides, carbides, fluorides, chlorides, chalcogenides, and metal alloys. Our results demonstrate that Ostwald's rule of stages can be exploited mechanistically at the molecular level to predictably access new metastable crystals from the amorphous phase in material synthesis.
AVIDa-hIL6: A Large-Scale VHH Dataset Produced from an Immunized Alpaca for Predicting Antigen-Antibody Interactions
Jun 06, 2023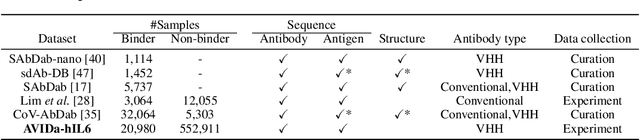
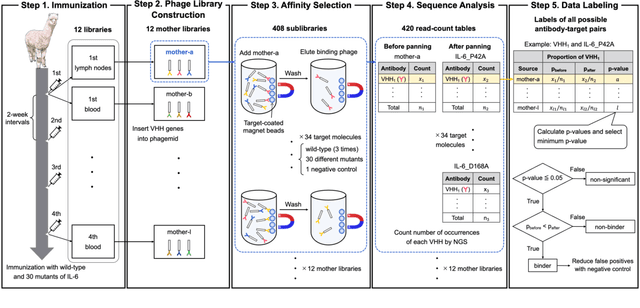
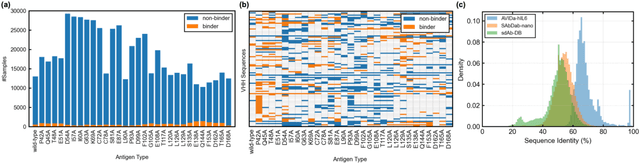

Antibodies have become an important class of therapeutic agents to treat human diseases. To accelerate therapeutic antibody discovery, computational methods, especially machine learning, have attracted considerable interest for predicting specific interactions between antibody candidates and target antigens such as viruses and bacteria. However, the publicly available datasets in existing works have notable limitations, such as small sizes and the lack of non-binding samples and exact amino acid sequences. To overcome these limitations, we have developed AVIDa-hIL6, a large-scale dataset for predicting antigen-antibody interactions in the variable domain of heavy chain of heavy chain antibodies (VHHs), produced from an alpaca immunized with the human interleukin-6 (IL-6) protein, as antigens. By leveraging the simple structure of VHHs, which facilitates identification of full-length amino acid sequences by DNA sequencing technology, AVIDa-hIL6 contains 573,891 antigen-VHH pairs with amino acid sequences. All the antigen-VHH pairs have reliable labels for binding or non-binding, as generated by a novel labeling method. Furthermore, via introduction of artificial mutations, AVIDa-hIL6 contains 30 different mutants in addition to wild-type IL-6 protein. This characteristic provides opportunities to develop machine learning models for predicting changes in antibody binding by antigen mutations. We report experimental benchmark results on AVIDa-hIL6 by using neural network-based baseline models. The results indicate that the existing models have potential, but further research is needed to generalize them to predict effective antibodies against unknown mutants. The dataset is available at https://avida-hil6.cognanous.com.
Machine Learning for Scent: Learning Generalizable Perceptual Representations of Small Molecules
Oct 25, 2019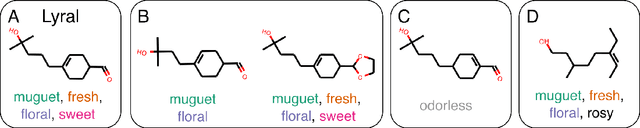
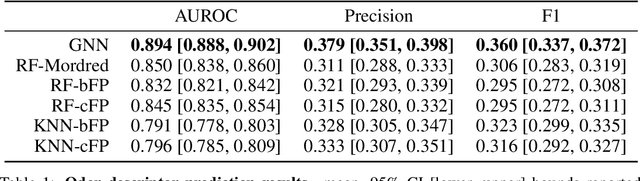
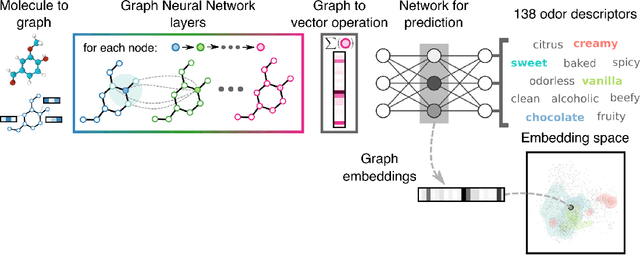
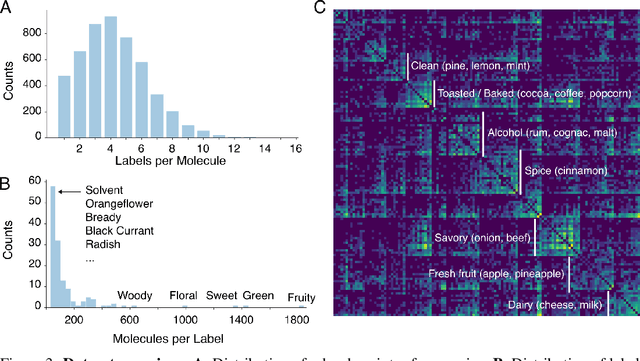
Predicting the relationship between a molecule's structure and its odor remains a difficult, decades-old task. This problem, termed quantitative structure-odor relationship (QSOR) modeling, is an important challenge in chemistry, impacting human nutrition, manufacture of synthetic fragrance, the environment, and sensory neuroscience. We propose the use of graph neural networks for QSOR, and show they significantly out-perform prior methods on a novel data set labeled by olfactory experts. Additional analysis shows that the learned embeddings from graph neural networks capture a meaningful odor space representation of the underlying relationship between structure and odor, as demonstrated by strong performance on two challenging transfer learning tasks. Machine learning has already had a large impact on the senses of sight and sound. Based on these early results with graph neural networks for molecular properties, we hope machine learning can eventually do for olfaction what it has already done for vision and hearing.
Predicting Electron-Ionization Mass Spectrometry using Neural Networks
Nov 21, 2018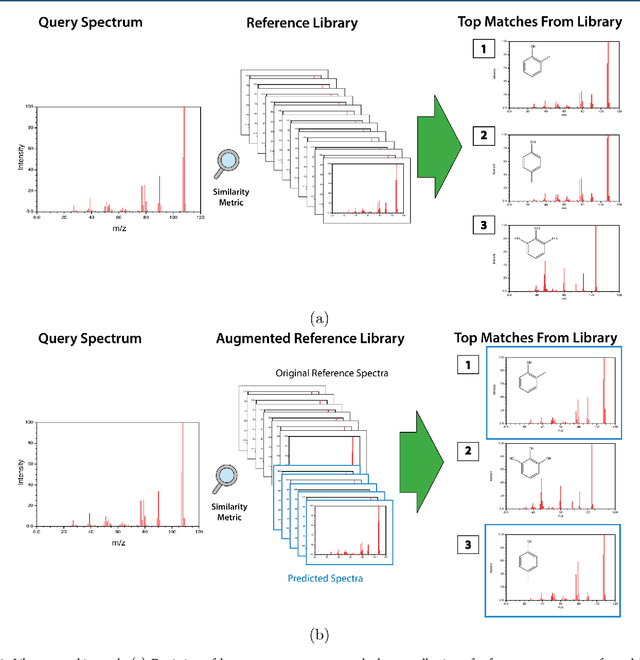
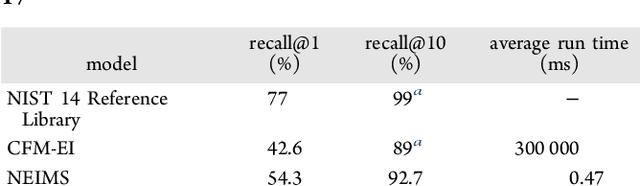
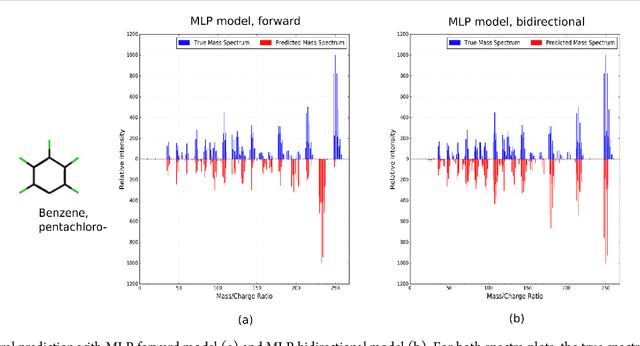
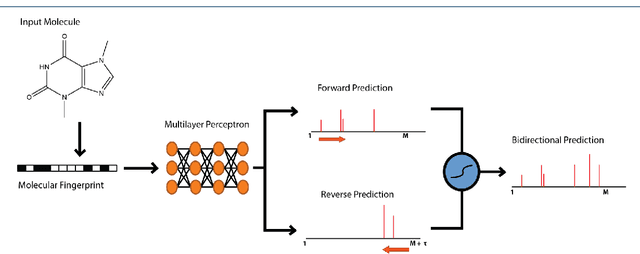
When confronted with a substance of unknown identity, researchers often perform mass spectrometry on the sample and compare the observed spectrum to a library of previously-collected spectra to identify the molecule. While popular, this approach will fail to identify molecules that are not in the existing library. In response, we propose to improve the library's coverage by augmenting it with synthetic spectra that are predicted using machine learning. We contribute a lightweight neural network model that quickly predicts mass spectra for small molecules. Achieving high accuracy predictions requires a novel neural network architecture that is designed to capture typical fragmentation patterns from electron ionization. We analyze the effects of our modeling innovations on library matching performance and compare our models to prior machine learning-based work on spectrum prediction.
Automatic chemical design using a data-driven continuous representation of molecules
Dec 05, 2017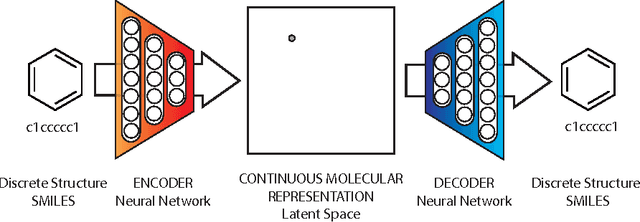
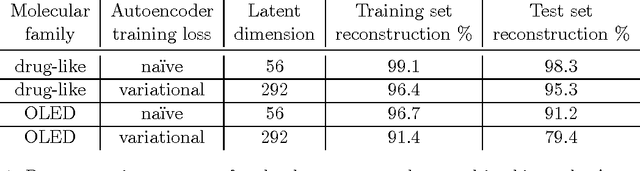
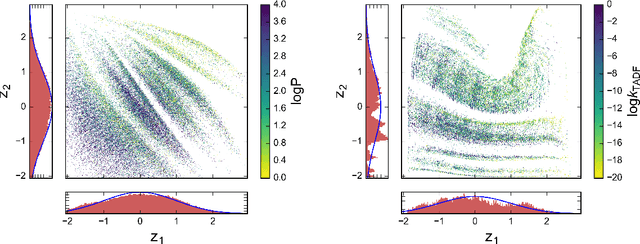
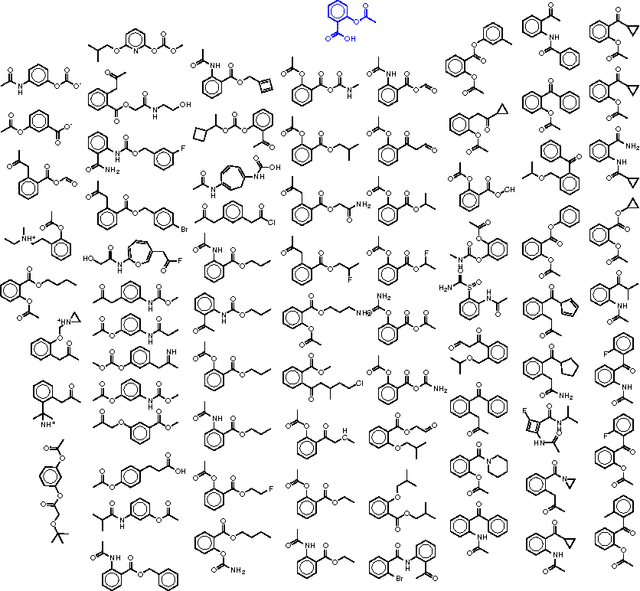
We report a method to convert discrete representations of molecules to and from a multidimensional continuous representation. This model allows us to generate new molecules for efficient exploration and optimization through open-ended spaces of chemical compounds. A deep neural network was trained on hundreds of thousands of existing chemical structures to construct three coupled functions: an encoder, a decoder and a predictor. The encoder converts the discrete representation of a molecule into a real-valued continuous vector, and the decoder converts these continuous vectors back to discrete molecular representations. The predictor estimates chemical properties from the latent continuous vector representation of the molecule. Continuous representations allow us to automatically generate novel chemical structures by performing simple operations in the latent space, such as decoding random vectors, perturbing known chemical structures, or interpolating between molecules. Continuous representations also allow the use of powerful gradient-based optimization to efficiently guide the search for optimized functional compounds. We demonstrate our method in the domain of drug-like molecules and also in the set of molecules with fewer that nine heavy atoms.
Neural networks for the prediction organic chemistry reactions
Oct 17, 2016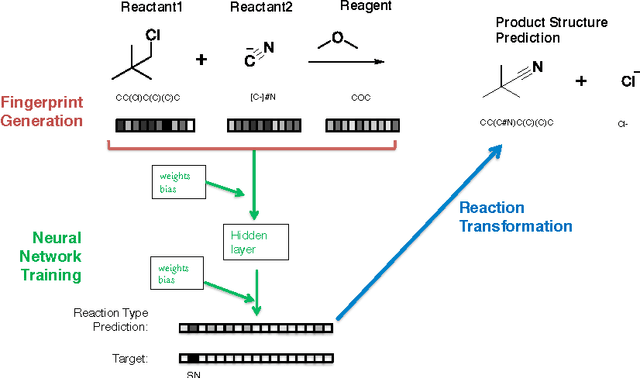

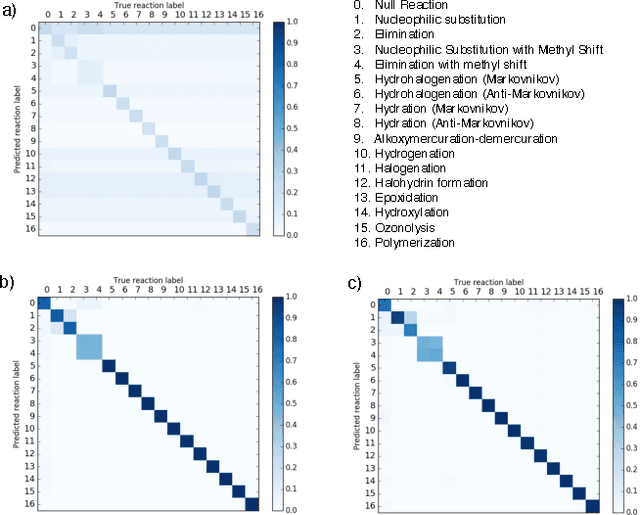
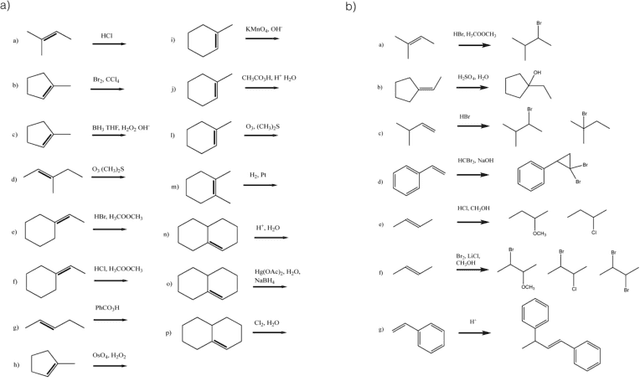
Reaction prediction remains one of the major challenges for organic chemistry, and is a pre-requisite for efficient synthetic planning. It is desirable to develop algorithms that, like humans, "learn" from being exposed to examples of the application of the rules of organic chemistry. We explore the use of neural networks for predicting reaction types, using a new reaction fingerprinting method. We combine this predictor with SMARTS transformations to build a system which, given a set of reagents and re- actants, predicts the likely products. We test this method on problems from a popular organic chemistry textbook.
* 21 pages, 5 figures