 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGenerative Hierarchical Materials Search
Sep 10, 2024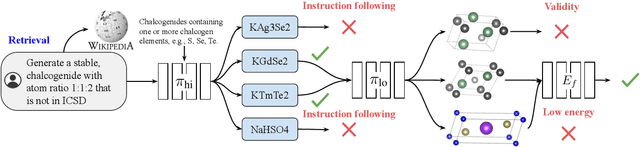

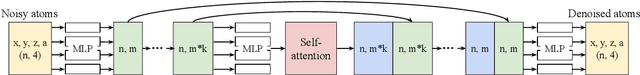

Generative models trained at scale can now produce text, video, and more recently, scientific data such as crystal structures. In applications of generative approaches to materials science, and in particular to crystal structures, the guidance from the domain expert in the form of high-level instructions can be essential for an automated system to output candidate crystals that are viable for downstream research. In this work, we formulate end-to-end language-to-structure generation as a multi-objective optimization problem, and propose Generative Hierarchical Materials Search (GenMS) for controllable generation of crystal structures. GenMS consists of (1) a language model that takes high-level natural language as input and generates intermediate textual information about a crystal (e.g., chemical formulae), and (2) a diffusion model that takes intermediate information as input and generates low-level continuous value crystal structures. GenMS additionally uses a graph neural network to predict properties (e.g., formation energy) from the generated crystal structures. During inference, GenMS leverages all three components to conduct a forward tree search over the space of possible structures. Experiments show that GenMS outperforms other alternatives of directly using language models to generate structures both in satisfying user request and in generating low-energy structures. We confirm that GenMS is able to generate common crystal structures such as double perovskites, or spinels, solely from natural language input, and hence can form the foundation for more complex structure generation in near future.
Predicting emergence of crystals from amorphous matter with deep learning
Oct 02, 2023Crystallization of the amorphous phases into metastable crystals plays a fundamental role in the formation of new matter, from geological to biological processes in nature to synthesis and development of new materials in the laboratory. Predicting the outcome of such phase transitions reliably would enable new research directions in these areas, but has remained beyond reach with molecular modeling or ab-initio methods. Here, we show that crystallization products of amorphous phases can be predicted in any inorganic chemistry by sampling the crystallization pathways of their local structural motifs at the atomistic level using universal deep learning potentials. We show that this approach identifies the crystal structures of polymorphs that initially nucleate from amorphous precursors with high accuracy across a diverse set of material systems, including polymorphic oxides, nitrides, carbides, fluorides, chlorides, chalcogenides, and metal alloys. Our results demonstrate that Ostwald's rule of stages can be exploited mechanistically at the molecular level to predictably access new metastable crystals from the amorphous phase in material synthesis.
Li$_x$CoO$_2$ phase stability studied by machine learning-enabled scale bridging between electronic structure, statistical mechanics and phase field theories
Apr 22, 2021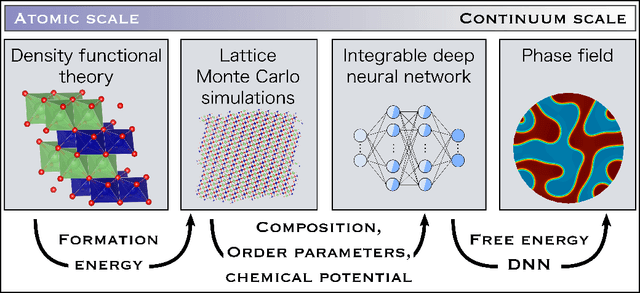
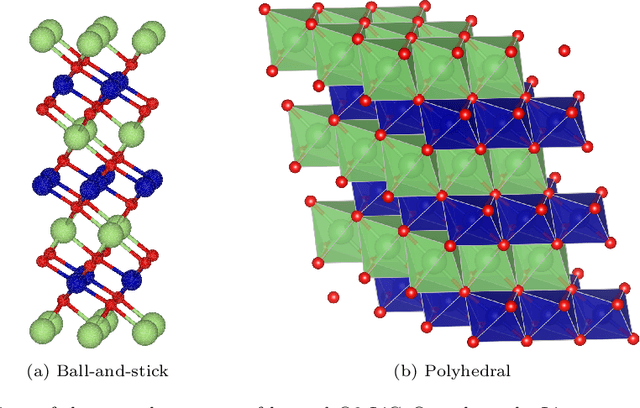
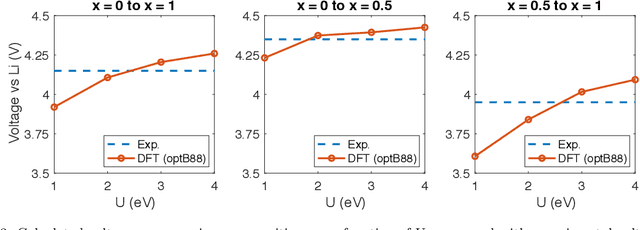
Li$_xTM$O$_2$ (TM={Ni, Co, Mn}) are promising cathodes for Li-ion batteries, whose electrochemical cycling performance is strongly governed by crystal structure and phase stability as a function of Li content at the atomistic scale. Here, we use Li$_x$CoO$_2$ (LCO) as a model system to benchmark a scale-bridging framework that combines density functional theory (DFT) calculations at the atomistic scale with phase field modeling at the continuum scale to understand the impact of phase stability on microstructure evolution. This scale bridging is accomplished by incorporating traditional statistical mechanics methods with integrable deep neural networks, which allows formation energies for specific atomic configurations to be coarse-grained and incorporated in a neural network description of the free energy of the material. The resulting realistic free energy functions enable atomistically informed phase-field simulations. These computational results allow us to make connections to experimental work on LCO cathode degradation as a function of temperature, morphology and particle size.