 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLi$_x$CoO$_2$ phase stability studied by machine learning-enabled scale bridging between electronic structure, statistical mechanics and phase field theories
Paper and Code
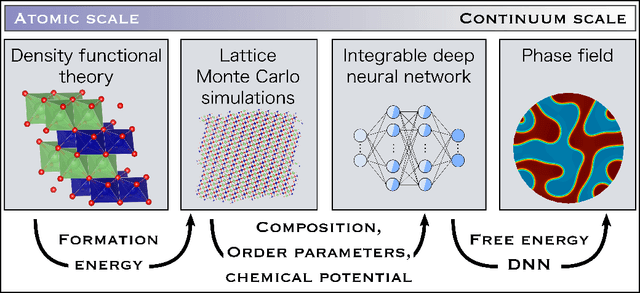
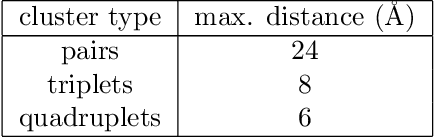
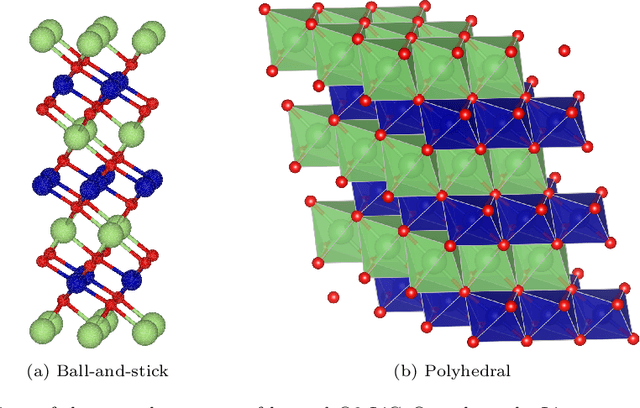
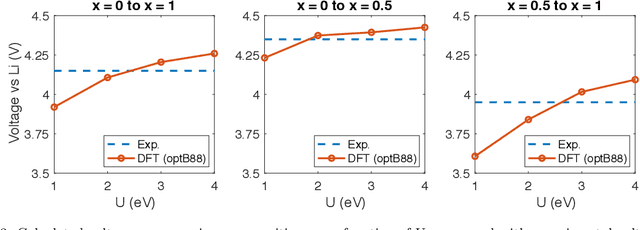
Li$_xTM$O$_2$ (TM={Ni, Co, Mn}) are promising cathodes for Li-ion batteries, whose electrochemical cycling performance is strongly governed by crystal structure and phase stability as a function of Li content at the atomistic scale. Here, we use Li$_x$CoO$_2$ (LCO) as a model system to benchmark a scale-bridging framework that combines density functional theory (DFT) calculations at the atomistic scale with phase field modeling at the continuum scale to understand the impact of phase stability on microstructure evolution. This scale bridging is accomplished by incorporating traditional statistical mechanics methods with integrable deep neural networks, which allows formation energies for specific atomic configurations to be coarse-grained and incorporated in a neural network description of the free energy of the material. The resulting realistic free energy functions enable atomistically informed phase-field simulations. These computational results allow us to make connections to experimental work on LCO cathode degradation as a function of temperature, morphology and particle size.