 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDiscrete Adjoint Schrödinger Bridge Sampler
Feb 09, 2026Learning discrete neural samplers is challenging due to the lack of gradients and combinatorial complexity. While stochastic optimal control (SOC) and Schrödinger bridge (SB) provide principled solutions, efficient SOC solvers like adjoint matching (AM), which excel in continuous domains, remain unexplored for discrete spaces. We bridge this gap by revealing that the core mechanism of AM is $\mathit{state}\text{-}\mathit{space~agnostic}$, and introduce $\mathbf{discrete~ASBS}$, a unified framework that extends AM and adjoint Schrödinger bridge sampler (ASBS) to discrete spaces. Theoretically, we analyze the optimality conditions of the discrete SB problem and its connection to SOC, identifying a necessary cyclic group structure on the state space to enable this extension. Empirically, discrete ASBS achieves competitive sample quality with significant advantages in training efficiency and scalability.
FragmentFlow: Scalable Transition State Generation for Large Molecules
Feb 02, 2026Transition states (TSs) are central to understanding and quantitatively predicting chemical reactivity and reaction mechanisms. Although traditional TS generation methods are computationally expensive, recent generative modeling approaches have enabled chemically meaningful TS prediction for relatively small molecules. However, these methods fail to generalize to practically relevant reaction substrates because of distribution shifts induced by increasing molecular sizes. Furthermore, TS geometries for larger molecules are not available at scale, making it infeasible to train generative models from scratch on such molecules. To address these challenges, we introduce FragmentFlow: a divide-and-conquer approach that trains a generative model to predict TS geometries for the reactive core atoms, which define the reaction mechanism. The full TS structure is then reconstructed by re-attaching substituent fragments to the predicted core. By operating on reactive cores, whose size and composition remain relatively invariant across molecular contexts, FragmentFlow mitigates distribution shifts in generative modeling. Evaluated on a new curated dataset of reactions involving reactants with up to 33 heavy atoms, FragmentFlow correctly identifies 90% of TSs while requiring 30% fewer saddle-point optimization steps than classical initialization schemes. These results point toward scalable TS generation for high-throughput reactivity studies.
Learning Collective Variables from Time-lagged Generation
Jul 10, 2025Rare events such as state transitions are difficult to observe directly with molecular dynamics simulations due to long timescales. Enhanced sampling techniques overcome this by introducing biases along carefully chosen low-dimensional features, known as collective variables (CVs), which capture the slow degrees of freedom. Machine learning approaches (MLCVs) have automated CV discovery, but existing methods typically focus on discriminating meta-stable states without fully encoding the detailed dynamics essential for accurate sampling. We propose TLC, a framework that learns CVs directly from time-lagged conditions of a generative model. Instead of modeling the static Boltzmann distribution, TLC models a time-lagged conditional distribution yielding CVs to capture the slow dynamic behavior. We validate TLC on the Alanine Dipeptide system using two CV-based enhanced sampling tasks: (i) steered molecular dynamics (SMD) and (ii) on-the-fly probability enhanced sampling (OPES), demonstrating equal or superior performance compared to existing MLCV methods in both transition path sampling and state discrimination.
Transferable Learning of Reaction Pathways from Geometric Priors
Apr 21, 2025



Identifying minimum-energy paths (MEPs) is crucial for understanding chemical reaction mechanisms but remains computationally demanding. We introduce MEPIN, a scalable machine-learning method for efficiently predicting MEPs from reactant and product configurations, without relying on transition-state geometries or pre-optimized reaction paths during training. The task is defined as predicting deviations from geometric interpolations along reaction coordinates. We address this task with a continuous reaction path model based on a symmetry-broken equivariant neural network that generates a flexible number of intermediate structures. The model is trained using an energy-based objective, with efficiency enhanced by incorporating geometric priors from geodesic interpolation as initial interpolations or pre-training objectives. Our approach generalizes across diverse chemical reactions and achieves accurate alignment with reference intrinsic reaction coordinates, as demonstrated on various small molecule reactions and [3+2] cycloadditions. Our method enables the exploration of large chemical reaction spaces with efficient, data-driven predictions of reaction pathways.
Symmetry-Constrained Generation of Diverse Low-Bandgap Molecules with Monte Carlo Tree Search
Oct 11, 2024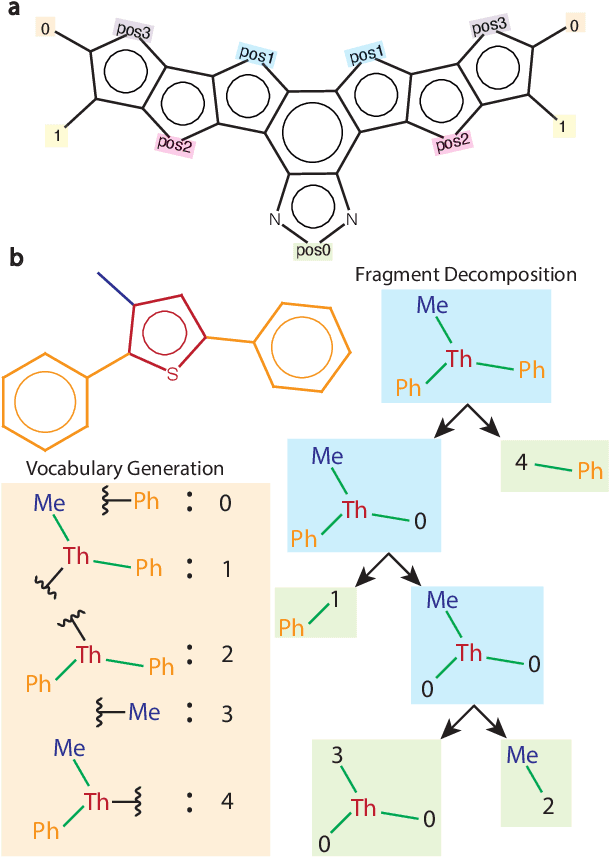
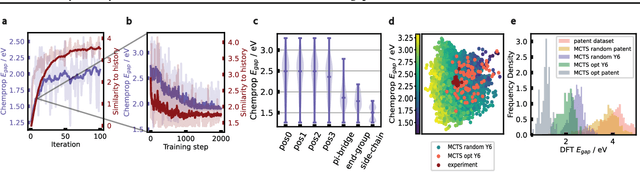
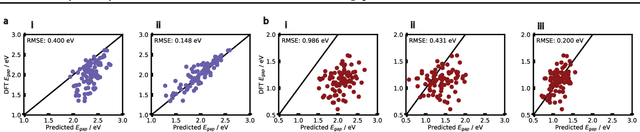
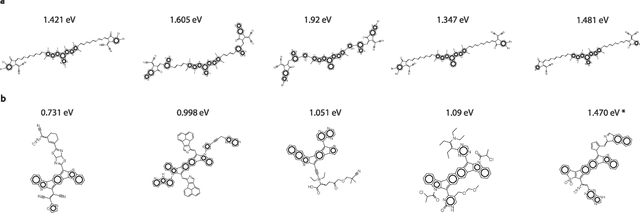
Organic optoelectronic materials are a promising avenue for next-generation electronic devices due to their solution processability, mechanical flexibility, and tunable electronic properties. In particular, near-infrared (NIR) sensitive molecules have unique applications in night-vision equipment and biomedical imaging. Molecular engineering has played a crucial role in developing non-fullerene acceptors (NFAs) such as the Y-series molecules, which have significantly improved the power conversion efficiency (PCE) of solar cells and enhanced spectral coverage in the NIR region. However, systematically designing molecules with targeted optoelectronic properties while ensuring synthetic accessibility remains a challenge. To address this, we leverage structural priors from domain-focused, patent-mined datasets of organic electronic molecules using a symmetry-aware fragment decomposition algorithm and a fragment-constrained Monte Carlo Tree Search (MCTS) generator. Our approach generates candidates that retain symmetry constraints from the patent dataset, while also exhibiting red-shifted absorption, as validated by TD-DFT calculations.
Efficient Generation of Molecular Clusters with Dual-Scale Equivariant Flow Matching
Oct 10, 2024Amorphous molecular solids offer a promising alternative to inorganic semiconductors, owing to their mechanical flexibility and solution processability. The packing structure of these materials plays a crucial role in determining their electronic and transport properties, which are key to enhancing the efficiency of devices like organic solar cells (OSCs). However, obtaining these optoelectronic properties computationally requires molecular dynamics (MD) simulations to generate a conformational ensemble, a process that can be computationally expensive due to the large system sizes involved. Recent advances have focused on using generative models, particularly flow-based models as Boltzmann generators, to improve the efficiency of MD sampling. In this work, we developed a dual-scale flow matching method that separates training and inference into coarse-grained and all-atom stages and enhances both the accuracy and efficiency of standard flow matching samplers. We demonstrate the effectiveness of this method on a dataset of Y6 molecular clusters obtained through MD simulations, and we benchmark its efficiency and accuracy against single-scale flow matching methods.
Think While You Generate: Discrete Diffusion with Planned Denoising
Oct 08, 2024



Discrete diffusion has achieved state-of-the-art performance, outperforming or approaching autoregressive models on standard benchmarks. In this work, we introduce Discrete Diffusion with Planned Denoising (DDPD), a novel framework that separates the generation process into two models: a planner and a denoiser. At inference time, the planner selects which positions to denoise next by identifying the most corrupted positions in need of denoising, including both initially corrupted and those requiring additional refinement. This plan-and-denoise approach enables more efficient reconstruction during generation by iteratively identifying and denoising corruptions in the optimal order. DDPD outperforms traditional denoiser-only mask diffusion methods, achieving superior results on language modeling benchmarks such as text8, OpenWebText, and token-based generation on ImageNet $256 \times 256$. Notably, in language modeling, DDPD significantly reduces the performance gap between diffusion-based and autoregressive methods in terms of generative perplexity. Code is available at https://github.com/liusulin/DDPD.
Flow Matching for Accelerated Simulation of Atomic Transport in Materials
Oct 02, 2024We introduce LiFlow, a generative framework to accelerate molecular dynamics (MD) simulations for crystalline materials that formulates the task as conditional generation of atomic displacements. The model uses flow matching, with a Propagator submodel to generate atomic displacements and a Corrector to locally correct unphysical geometries, and incorporates an adaptive prior based on the Maxwell-Boltzmann distribution to account for chemical and thermal conditions. We benchmark LiFlow on a dataset comprising 25-ps trajectories of lithium diffusion across 4,186 solid-state electrolyte (SSE) candidates at four temperatures. The model obtains a consistent Spearman rank correlation of 0.7-0.8 for lithium mean squared displacement (MSD) predictions on unseen compositions. Furthermore, LiFlow generalizes from short training trajectories to larger supercells and longer simulations while maintaining high accuracy. With speed-ups of up to 600,000$\times$ compared to first-principles methods, LiFlow enables scalable simulations at significantly larger length and time scales.
Learning Ordering in Crystalline Materials with Symmetry-Aware Graph Neural Networks
Sep 20, 2024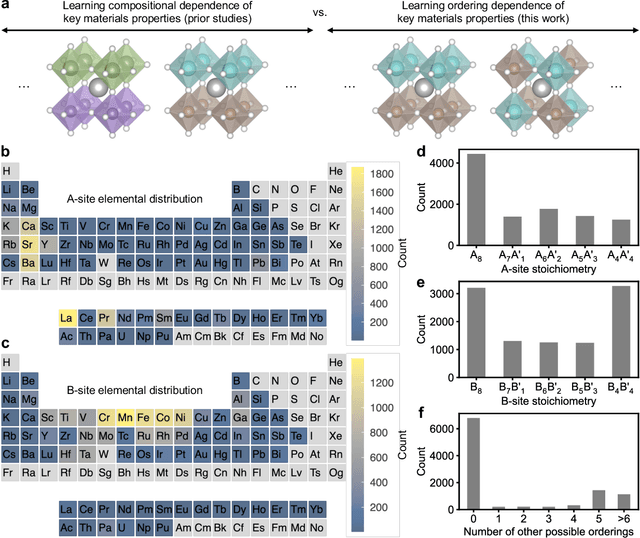
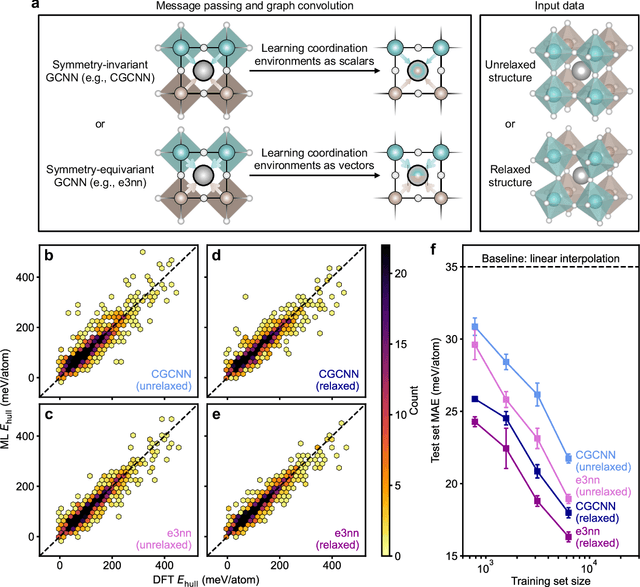
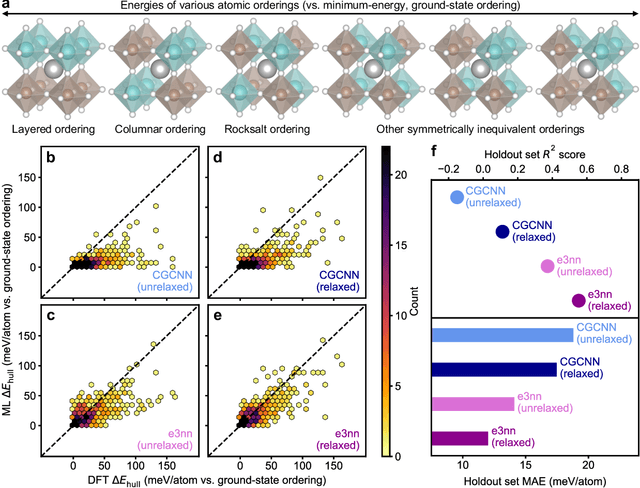
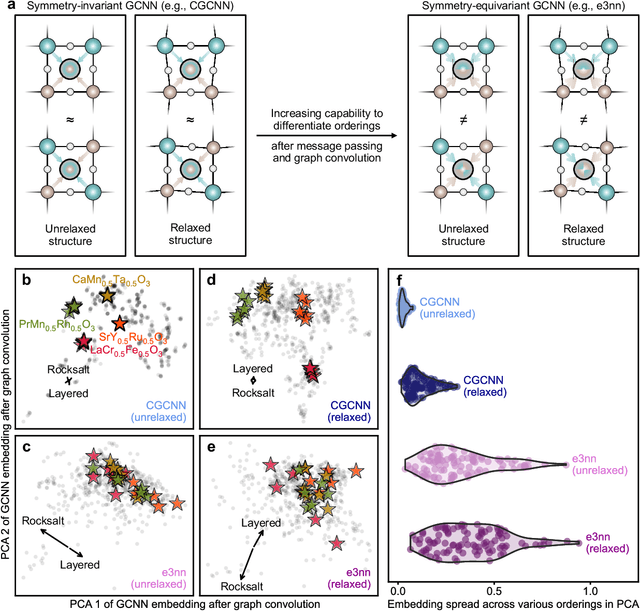
Graph convolutional neural networks (GCNNs) have become a machine learning workhorse for screening the chemical space of crystalline materials in fields such as catalysis and energy storage, by predicting properties from structures. Multicomponent materials, however, present a unique challenge since they can exhibit chemical (dis)order, where a given lattice structure can encompass a variety of elemental arrangements ranging from highly ordered structures to fully disordered solid solutions. Critically, properties like stability, strength, and catalytic performance depend not only on structures but also on orderings. To enable rigorous materials design, it is thus critical to ensure GCNNs are capable of distinguishing among atomic orderings. However, the ordering-aware capability of GCNNs has been poorly understood. Here, we benchmark various neural network architectures for capturing the ordering-dependent energetics of multicomponent materials in a custom-made dataset generated with high-throughput atomistic simulations. Conventional symmetry-invariant GCNNs were found unable to discern the structural difference between the diverse symmetrically inequivalent atomic orderings of the same material, while symmetry-equivariant model architectures could inherently preserve and differentiate the distinct crystallographic symmetries of various orderings.
Interpolation and differentiation of alchemical degrees of freedom in machine learning interatomic potentials
Apr 16, 2024Machine learning interatomic potentials (MLIPs) have become a workhorse of modern atomistic simulations, and recently published universal MLIPs, pre-trained on large datasets, have demonstrated remarkable accuracy and generalizability. However, the computational cost of MLIPs limits their applicability to chemically disordered systems requiring large simulation cells or to sample-intensive statistical methods. Here, we report the use of continuous and differentiable alchemical degrees of freedom in atomistic materials simulations, exploiting the fact that graph neural network MLIPs represent discrete elements as real-valued tensors. The proposed method introduces alchemical atoms with corresponding weights into the input graph, alongside modifications to the message-passing and readout mechanisms of MLIPs, and allows smooth interpolation between the compositional states of materials. The end-to-end differentiability of MLIPs enables efficient calculation of the gradient of energy with respect to the compositional weights. Leveraging these gradients, we propose methodologies for optimizing the composition of solid solutions towards target macroscopic properties and conducting alchemical free energy simulations to quantify the free energy of vacancy formation and composition changes. The approach offers an avenue for extending the capabilities of universal MLIPs in the modeling of compositional disorder and characterizing the phase stabilities of complex materials systems.