 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFlow Matching for Accelerated Simulation of Atomic Transport in Materials
Oct 02, 2024We introduce LiFlow, a generative framework to accelerate molecular dynamics (MD) simulations for crystalline materials that formulates the task as conditional generation of atomic displacements. The model uses flow matching, with a Propagator submodel to generate atomic displacements and a Corrector to locally correct unphysical geometries, and incorporates an adaptive prior based on the Maxwell-Boltzmann distribution to account for chemical and thermal conditions. We benchmark LiFlow on a dataset comprising 25-ps trajectories of lithium diffusion across 4,186 solid-state electrolyte (SSE) candidates at four temperatures. The model obtains a consistent Spearman rank correlation of 0.7-0.8 for lithium mean squared displacement (MSD) predictions on unseen compositions. Furthermore, LiFlow generalizes from short training trajectories to larger supercells and longer simulations while maintaining high accuracy. With speed-ups of up to 600,000$\times$ compared to first-principles methods, LiFlow enables scalable simulations at significantly larger length and time scales.
Overcoming systematic softening in universal machine learning interatomic potentials by fine-tuning
May 11, 2024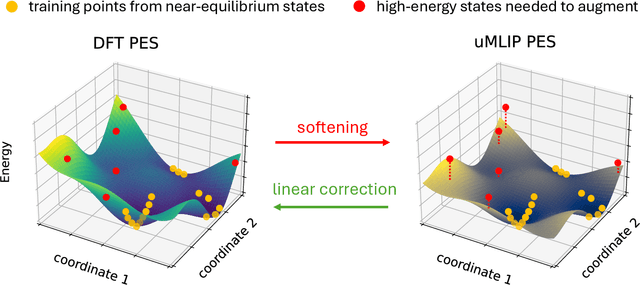
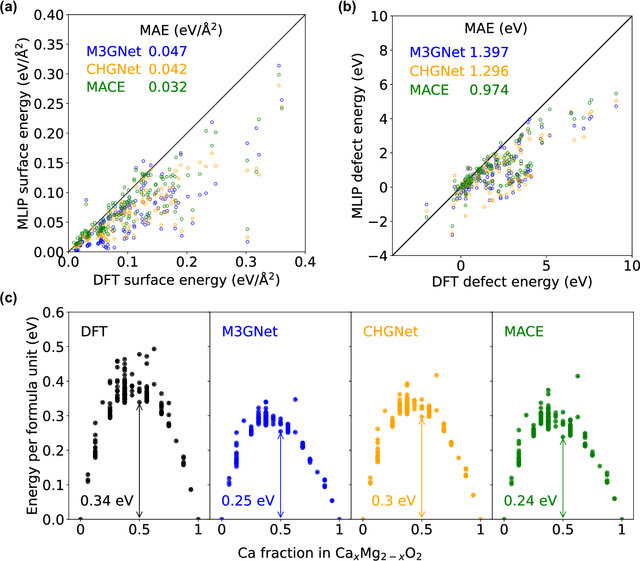
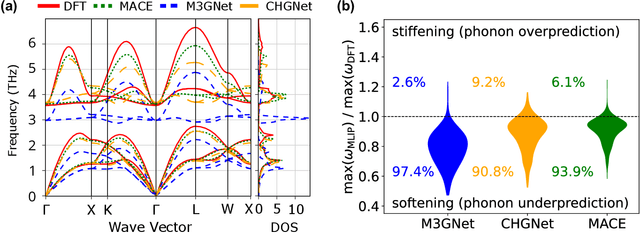
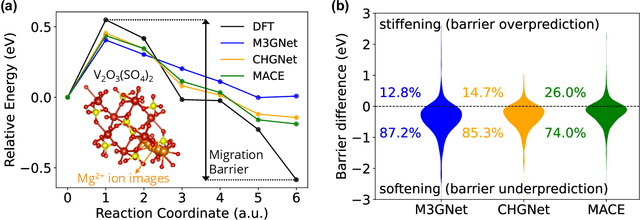
Machine learning interatomic potentials (MLIPs) have introduced a new paradigm for atomic simulations. Recent advancements have seen the emergence of universal MLIPs (uMLIPs) that are pre-trained on diverse materials datasets, providing opportunities for both ready-to-use universal force fields and robust foundations for downstream machine learning refinements. However, their performance in extrapolating to out-of-distribution complex atomic environments remains unclear. In this study, we highlight a consistent potential energy surface (PES) softening effect in three uMLIPs: M3GNet, CHGNet, and MACE-MP-0, which is characterized by energy and force under-prediction in a series of atomic-modeling benchmarks including surfaces, defects, solid-solution energetics, phonon vibration modes, ion migration barriers, and general high-energy states. We find that the PES softening behavior originates from a systematic underprediction error of the PES curvature, which derives from the biased sampling of near-equilibrium atomic arrangements in uMLIP pre-training datasets. We demonstrate that the PES softening issue can be effectively rectified by fine-tuning with a single additional data point. Our findings suggest that a considerable fraction of uMLIP errors are highly systematic, and can therefore be efficiently corrected. This result rationalizes the data-efficient fine-tuning performance boost commonly observed with foundational MLIPs. We argue for the importance of a comprehensive materials dataset with improved PES sampling for next-generation foundational MLIPs.
CHGNet: Pretrained universal neural network potential for charge-informed atomistic modeling
Feb 28, 2023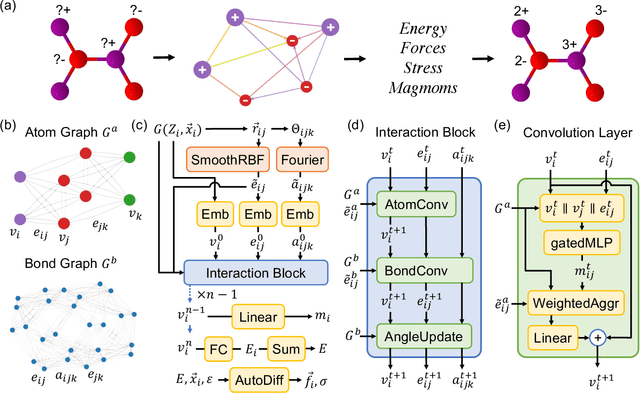
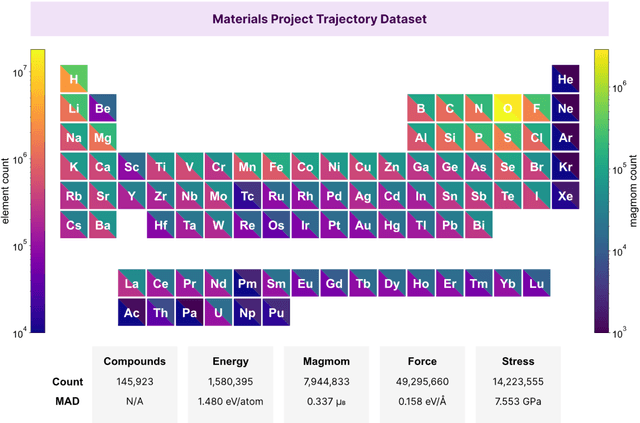
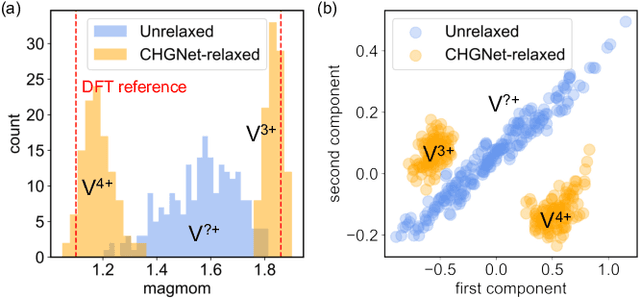
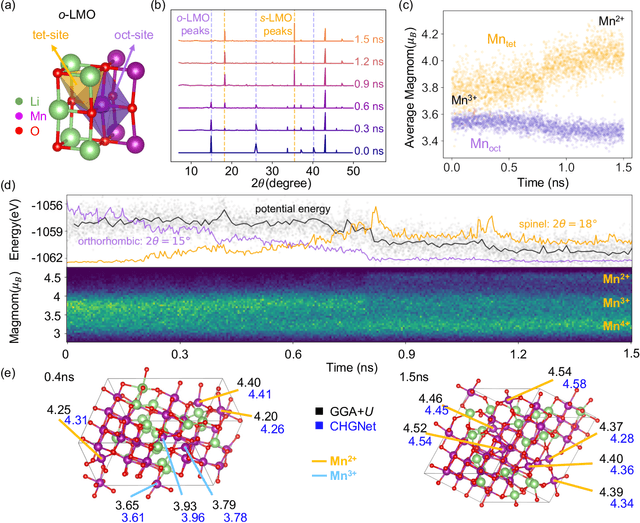
The simulation of large-scale systems with complex electron interactions remains one of the greatest challenges for the atomistic modeling of materials. Although classical force-fields often fail to describe the coupling between electronic states and ionic rearrangements, the more accurate \textit{ab-initio} molecular dynamics suffers from computational complexity that prevents long-time and large-scale simulations, which are essential to study many technologically relevant phenomena, such as reactions, ion migrations, phase transformations, and degradation. In this work, we present the Crystal Hamiltonian Graph neural Network (CHGNet) as a novel machine-learning interatomic potential (MLIP), using a graph-neural-network-based force-field to model a universal potential energy surface. CHGNet is pretrained on the energies, forces, stresses, and magnetic moments from the Materials Project Trajectory Dataset, which consists of over 10 years of density functional theory static and relaxation trajectories of $\sim 1.5$ million inorganic structures. The explicit inclusion of magnetic moments enables CHGNet to learn and accurately represent the orbital occupancy of electrons, enhancing its capability to describe both atomic and electronic degrees of freedom. We demonstrate several applications of CHGNet in solid-state materials, including charge-informed molecular dynamics in Li$_x$MnO$_2$, the finite temperature phase diagram for Li$_x$FePO$_4$ and Li diffusion in garnet conductors. We critically analyze the significance of including charge information for capturing appropriate chemistry, and we provide new insights into ionic systems with additional electronic degrees of freedom that can not be observed by previous MLIPs.