 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGeneralized Schrödinger Bridge on Graphs
Feb 04, 2026Transportation on graphs is a fundamental challenge across many domains, where decisions must respect topological and operational constraints. Despite the need for actionable policies, existing graph-transport methods lack this expressivity. They rely on restrictive assumptions, fail to generalize across sparse topologies, and scale poorly with graph size and time horizon. To address these issues, we introduce Generalized Schrödinger Bridge on Graphs (GSBoG), a novel scalable data-driven framework for learning executable controlled continuous-time Markov chain (CTMC) policies on arbitrary graphs under state cost augmented dynamics. Notably, GSBoG learns trajectory-level policies, avoiding dense global solvers and thereby enhancing scalability. This is achieved via a likelihood optimization approach, satisfying the endpoint marginals, while simultaneously optimizing intermediate behavior under state-dependent running costs. Extensive experimentation on challenging real-world graph topologies shows that GSBoG reliably learns accurate, topology-respecting policies while optimizing application-specific intermediate state costs, highlighting its broad applicability and paving new avenues for cost-aware dynamical transport on general graphs.
FragmentFlow: Scalable Transition State Generation for Large Molecules
Feb 02, 2026Transition states (TSs) are central to understanding and quantitatively predicting chemical reactivity and reaction mechanisms. Although traditional TS generation methods are computationally expensive, recent generative modeling approaches have enabled chemically meaningful TS prediction for relatively small molecules. However, these methods fail to generalize to practically relevant reaction substrates because of distribution shifts induced by increasing molecular sizes. Furthermore, TS geometries for larger molecules are not available at scale, making it infeasible to train generative models from scratch on such molecules. To address these challenges, we introduce FragmentFlow: a divide-and-conquer approach that trains a generative model to predict TS geometries for the reactive core atoms, which define the reaction mechanism. The full TS structure is then reconstructed by re-attaching substituent fragments to the predicted core. By operating on reactive cores, whose size and composition remain relatively invariant across molecular contexts, FragmentFlow mitigates distribution shifts in generative modeling. Evaluated on a new curated dataset of reactions involving reactants with up to 33 heavy atoms, FragmentFlow correctly identifies 90% of TSs while requiring 30% fewer saddle-point optimization steps than classical initialization schemes. These results point toward scalable TS generation for high-throughput reactivity studies.
Transferable Learning of Reaction Pathways from Geometric Priors
Apr 21, 2025



Identifying minimum-energy paths (MEPs) is crucial for understanding chemical reaction mechanisms but remains computationally demanding. We introduce MEPIN, a scalable machine-learning method for efficiently predicting MEPs from reactant and product configurations, without relying on transition-state geometries or pre-optimized reaction paths during training. The task is defined as predicting deviations from geometric interpolations along reaction coordinates. We address this task with a continuous reaction path model based on a symmetry-broken equivariant neural network that generates a flexible number of intermediate structures. The model is trained using an energy-based objective, with efficiency enhanced by incorporating geometric priors from geodesic interpolation as initial interpolations or pre-training objectives. Our approach generalizes across diverse chemical reactions and achieves accurate alignment with reference intrinsic reaction coordinates, as demonstrated on various small molecule reactions and [3+2] cycloadditions. Our method enables the exploration of large chemical reaction spaces with efficient, data-driven predictions of reaction pathways.
Symmetry-Constrained Generation of Diverse Low-Bandgap Molecules with Monte Carlo Tree Search
Oct 11, 2024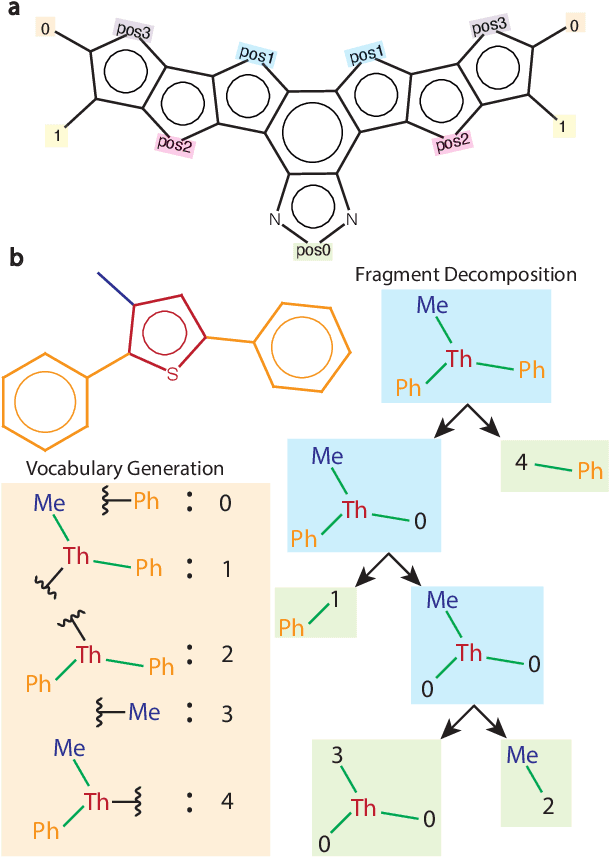
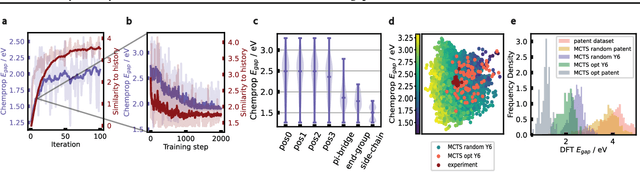
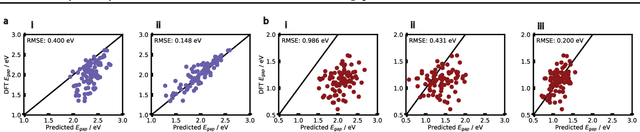
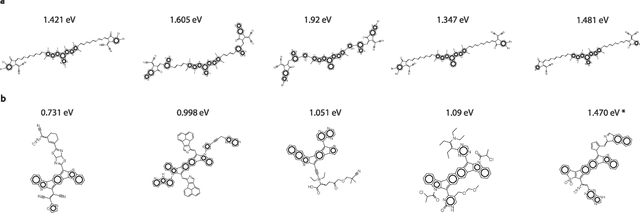
Organic optoelectronic materials are a promising avenue for next-generation electronic devices due to their solution processability, mechanical flexibility, and tunable electronic properties. In particular, near-infrared (NIR) sensitive molecules have unique applications in night-vision equipment and biomedical imaging. Molecular engineering has played a crucial role in developing non-fullerene acceptors (NFAs) such as the Y-series molecules, which have significantly improved the power conversion efficiency (PCE) of solar cells and enhanced spectral coverage in the NIR region. However, systematically designing molecules with targeted optoelectronic properties while ensuring synthetic accessibility remains a challenge. To address this, we leverage structural priors from domain-focused, patent-mined datasets of organic electronic molecules using a symmetry-aware fragment decomposition algorithm and a fragment-constrained Monte Carlo Tree Search (MCTS) generator. Our approach generates candidates that retain symmetry constraints from the patent dataset, while also exhibiting red-shifted absorption, as validated by TD-DFT calculations.
Think While You Generate: Discrete Diffusion with Planned Denoising
Oct 08, 2024



Discrete diffusion has achieved state-of-the-art performance, outperforming or approaching autoregressive models on standard benchmarks. In this work, we introduce Discrete Diffusion with Planned Denoising (DDPD), a novel framework that separates the generation process into two models: a planner and a denoiser. At inference time, the planner selects which positions to denoise next by identifying the most corrupted positions in need of denoising, including both initially corrupted and those requiring additional refinement. This plan-and-denoise approach enables more efficient reconstruction during generation by iteratively identifying and denoising corruptions in the optimal order. DDPD outperforms traditional denoiser-only mask diffusion methods, achieving superior results on language modeling benchmarks such as text8, OpenWebText, and token-based generation on ImageNet $256 \times 256$. Notably, in language modeling, DDPD significantly reduces the performance gap between diffusion-based and autoregressive methods in terms of generative perplexity. Code is available at https://github.com/liusulin/DDPD.
Flow Matching for Accelerated Simulation of Atomic Transport in Materials
Oct 02, 2024We introduce LiFlow, a generative framework to accelerate molecular dynamics (MD) simulations for crystalline materials that formulates the task as conditional generation of atomic displacements. The model uses flow matching, with a Propagator submodel to generate atomic displacements and a Corrector to locally correct unphysical geometries, and incorporates an adaptive prior based on the Maxwell-Boltzmann distribution to account for chemical and thermal conditions. We benchmark LiFlow on a dataset comprising 25-ps trajectories of lithium diffusion across 4,186 solid-state electrolyte (SSE) candidates at four temperatures. The model obtains a consistent Spearman rank correlation of 0.7-0.8 for lithium mean squared displacement (MSD) predictions on unseen compositions. Furthermore, LiFlow generalizes from short training trajectories to larger supercells and longer simulations while maintaining high accuracy. With speed-ups of up to 600,000$\times$ compared to first-principles methods, LiFlow enables scalable simulations at significantly larger length and time scales.
Understanding active learning of molecular docking and its applications
Jun 14, 2024With the advancing capabilities of computational methodologies and resources, ultra-large-scale virtual screening via molecular docking has emerged as a prominent strategy for in silico hit discovery. Given the exhaustive nature of ultra-large-scale virtual screening, active learning methodologies have garnered attention as a means to mitigate computational cost through iterative small-scale docking and machine learning model training. While the efficacy of active learning methodologies has been empirically validated in extant literature, a critical investigation remains in how surrogate models can predict docking score without considering three-dimensional structural features, such as receptor conformation and binding poses. In this paper, we thus investigate how active learning methodologies effectively predict docking scores using only 2D structures and under what circumstances they may work particularly well through benchmark studies encompassing six receptor targets. Our findings suggest that surrogate models tend to memorize structural patterns prevalent in high docking scored compounds obtained during acquisition steps. Despite this tendency, surrogate models demonstrate utility in virtual screening, as exemplified in the identification of actives from DUD-E dataset and high docking-scored compounds from EnamineReal library, a significantly larger set than the initial screening pool. Our comprehensive analysis underscores the reliability and potential applicability of active learning methodologies in virtual screening campaigns.
Interpolation and differentiation of alchemical degrees of freedom in machine learning interatomic potentials
Apr 16, 2024Machine learning interatomic potentials (MLIPs) have become a workhorse of modern atomistic simulations, and recently published universal MLIPs, pre-trained on large datasets, have demonstrated remarkable accuracy and generalizability. However, the computational cost of MLIPs limits their applicability to chemically disordered systems requiring large simulation cells or to sample-intensive statistical methods. Here, we report the use of continuous and differentiable alchemical degrees of freedom in atomistic materials simulations, exploiting the fact that graph neural network MLIPs represent discrete elements as real-valued tensors. The proposed method introduces alchemical atoms with corresponding weights into the input graph, alongside modifications to the message-passing and readout mechanisms of MLIPs, and allows smooth interpolation between the compositional states of materials. The end-to-end differentiability of MLIPs enables efficient calculation of the gradient of energy with respect to the compositional weights. Leveraging these gradients, we propose methodologies for optimizing the composition of solid solutions towards target macroscopic properties and conducting alchemical free energy simulations to quantify the free energy of vacancy formation and composition changes. The approach offers an avenue for extending the capabilities of universal MLIPs in the modeling of compositional disorder and characterizing the phase stabilities of complex materials systems.
Learning Collective Variables for Protein Folding with Labeled Data Augmentation through Geodesic Interpolation
Feb 02, 2024



In molecular dynamics (MD) simulations, rare events, such as protein folding, are typically studied by means of enhanced sampling techniques, most of which rely on the definition of a collective variable (CV) along which the acceleration occurs. Obtaining an expressive CV is crucial, but often hindered by the lack of information about the particular event, e.g., the transition from unfolded to folded conformation. We propose a simulation-free data augmentation strategy using physics-inspired metrics to generate geodesic interpolations resembling protein folding transitions, thereby improving sampling efficiency without true transition state samples. Leveraging interpolation progress parameters, we introduce a regression-based learning scheme for CV models, which outperforms classifier-based methods when transition state data is limited and noisy
Linking the Neural Machine Translation and the Prediction of Organic Chemistry Reactions
Dec 29, 2016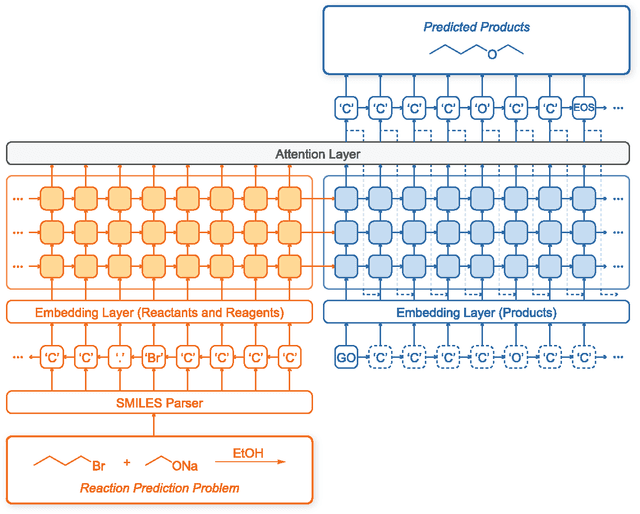



Finding the main product of a chemical reaction is one of the important problems of organic chemistry. This paper describes a method of applying a neural machine translation model to the prediction of organic chemical reactions. In order to translate 'reactants and reagents' to 'products', a gated recurrent unit based sequence-to-sequence model and a parser to generate input tokens for model from reaction SMILES strings were built. Training sets are composed of reactions from the patent databases, and reactions manually generated applying the elementary reactions in an organic chemistry textbook of Wade. The trained models were tested by examples and problems in the textbook. The prediction process does not need manual encoding of rules (e.g., SMARTS transformations) to predict products, hence it only needs sufficient training reaction sets to learn new types of reactions.