 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUnderstanding active learning of molecular docking and its applications
Jun 14, 2024



With the advancing capabilities of computational methodologies and resources, ultra-large-scale virtual screening via molecular docking has emerged as a prominent strategy for in silico hit discovery. Given the exhaustive nature of ultra-large-scale virtual screening, active learning methodologies have garnered attention as a means to mitigate computational cost through iterative small-scale docking and machine learning model training. While the efficacy of active learning methodologies has been empirically validated in extant literature, a critical investigation remains in how surrogate models can predict docking score without considering three-dimensional structural features, such as receptor conformation and binding poses. In this paper, we thus investigate how active learning methodologies effectively predict docking scores using only 2D structures and under what circumstances they may work particularly well through benchmark studies encompassing six receptor targets. Our findings suggest that surrogate models tend to memorize structural patterns prevalent in high docking scored compounds obtained during acquisition steps. Despite this tendency, surrogate models demonstrate utility in virtual screening, as exemplified in the identification of actives from DUD-E dataset and high docking-scored compounds from EnamineReal library, a significantly larger set than the initial screening pool. Our comprehensive analysis underscores the reliability and potential applicability of active learning methodologies in virtual screening campaigns.
Hit and Lead Discovery with Explorative RL and Fragment-based Molecule Generation
Oct 05, 2021
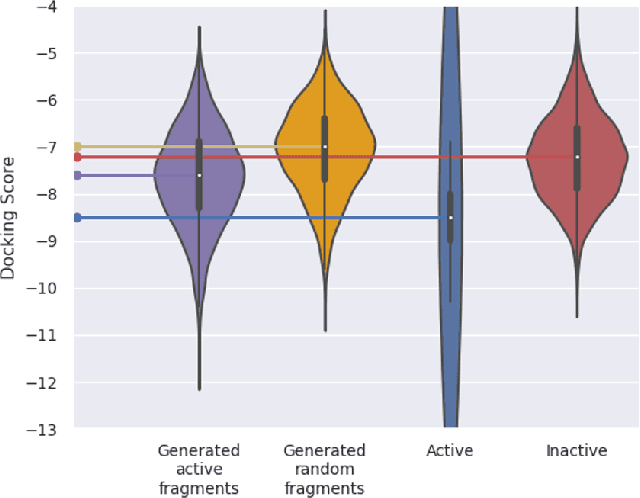

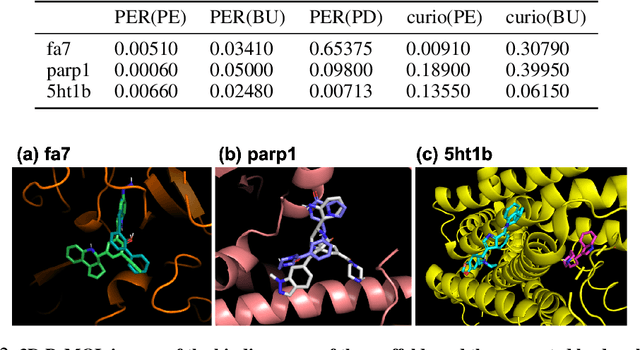
Recently, utilizing reinforcement learning (RL) to generate molecules with desired properties has been highlighted as a promising strategy for drug design. A molecular docking program - a physical simulation that estimates protein-small molecule binding affinity - can be an ideal reward scoring function for RL, as it is a straightforward proxy of the therapeutic potential. Still, two imminent challenges exist for this task. First, the models often fail to generate chemically realistic and pharmacochemically acceptable molecules. Second, the docking score optimization is a difficult exploration problem that involves many local optima and less smooth surfaces with respect to molecular structure. To tackle these challenges, we propose a novel RL framework that generates pharmacochemically acceptable molecules with large docking scores. Our method - Fragment-based generative RL with Explorative Experience replay for Drug design (FREED) - constrains the generated molecules to a realistic and qualified chemical space and effectively explores the space to find drugs by coupling our fragment-based generation method and a novel error-prioritized experience replay (PER). We also show that our model performs well on both de novo and scaffold-based schemes. Our model produces molecules of higher quality compared to existing methods while achieving state-of-the-art performance on two of three targets in terms of the docking scores of the generated molecules. We further show with ablation studies that our method, predictive error-PER (FREED(PE)), significantly improves the model performance.
A benchmark study on reliable molecular supervised learning via Bayesian learning
Jul 01, 2020

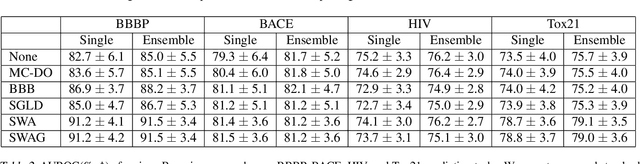

Virtual screening aims to find desirable compounds from chemical library by using computational methods. For this purpose with machine learning, model outputs that can be interpreted as predictive probability will be beneficial, in that a high prediction score corresponds to high probability of correctness. In this work, we present a study on the prediction performance and reliability of graph neural networks trained with the recently proposed Bayesian learning algorithms. Our work shows that Bayesian learning algorithms allow well-calibrated predictions for various GNN architectures and classification tasks. Also, we show the implications of reliable predictions on virtual screening, where Bayesian learning may lead to higher success in finding hit compounds.
A comprehensive study on the prediction reliability of graph neural networks for virtual screening
Mar 17, 2020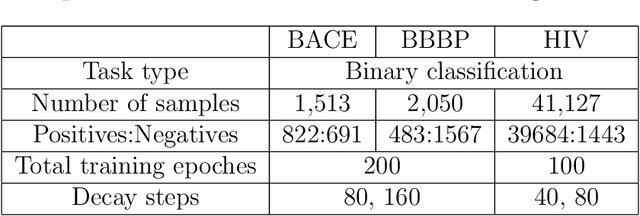
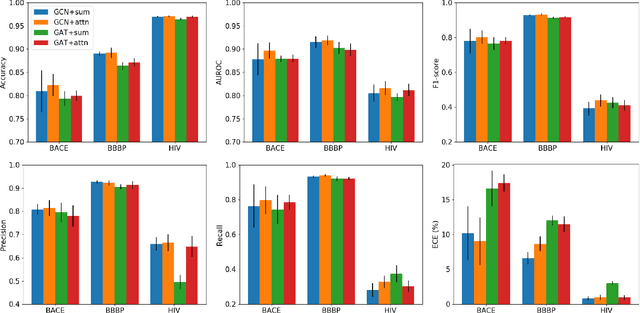
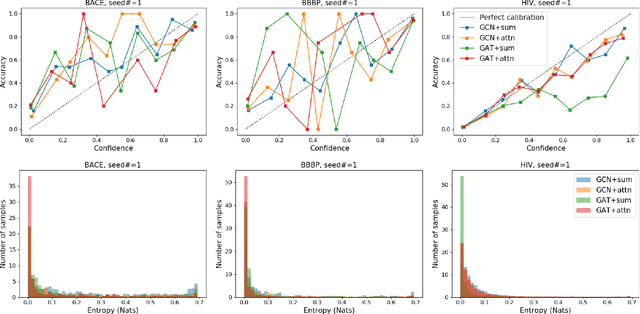
Prediction models based on deep neural networks are increasingly gaining attention for fast and accurate virtual screening systems. For decision makings in virtual screening, researchers find it useful to interpret an output of classification system as probability, since such interpretation allows them to filter out more desirable compounds. However, probabilistic interpretation cannot be correct for models that hold over-parameterization problems or inappropriate regularizations, leading to unreliable prediction and decision making. In this regard, we concern the reliability of neural prediction models on molecular properties, especially when models are trained with sparse data points and imbalanced distributions. This work aims to propose guidelines for training reliable models, we thus provide methodological details and ablation studies on the following train principles. We investigate the effects of model architectures, regularization methods, and loss functions on the prediction performance and reliability of classification results. Moreover, we evaluate prediction reliability of models on virtual screening scenario. Our result highlights that correct choice of regularization and inference methods is evidently important to achieve high success rate, especially in data imbalanced situation. All experiments were performed under a single unified model implementation to alleviate external randomness in model training and to enable precise comparison of results.
Molecular Generative Model Based On Adversarially Regularized Autoencoder
Nov 13, 2019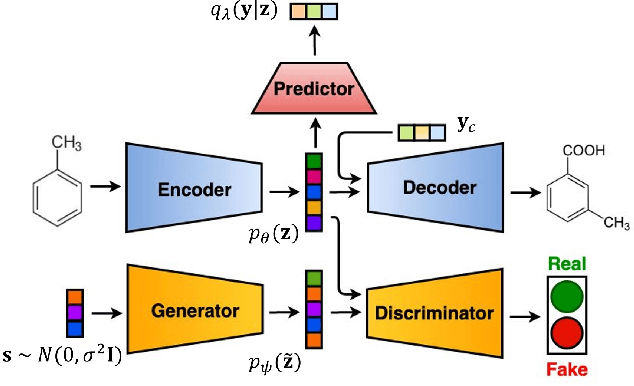
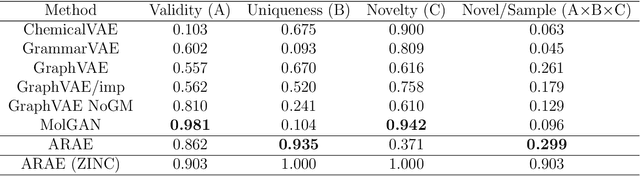
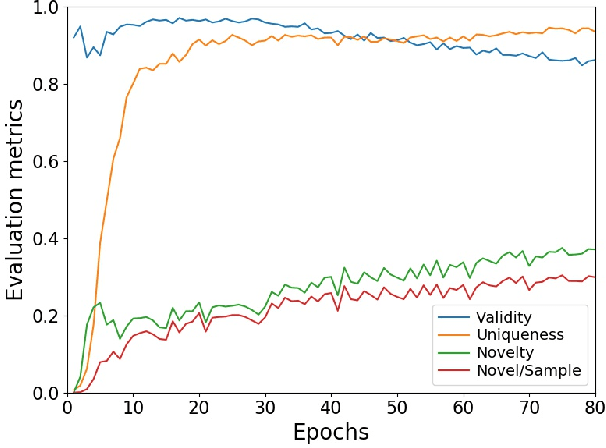
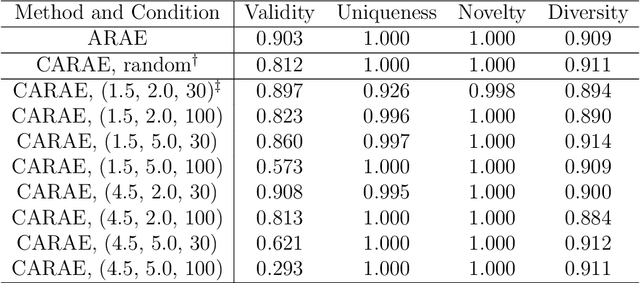
Deep generative models are attracting great attention as a new promising approach for molecular design. All models reported so far are based on either variational autoencoder (VAE) or generative adversarial network (GAN). Here we propose a new type model based on an adversarially regularized autoencoder (ARAE). It basically uses latent variables like VAE, but the distribution of the latent variables is obtained by adversarial training like in GAN. The latter is intended to avoid both inappropriate approximation of posterior distribution in VAE and difficulty in handling discrete variables in GAN. Our benchmark study showed that ARAE indeed outperformed conventional models in terms of validity, uniqueness, and novelty per generated molecule. We also demonstrated successful conditional generation of drug-like molecules with ARAE for both cases of single and multiple properties control. As a potential real-world application, we could generate EGFR inhibitors sharing the scaffolds of known active molecules while satisfying drug-like conditions simultaneously.
Predicting drug-target interaction using 3D structure-embedded graph representations from graph neural networks
Apr 17, 2019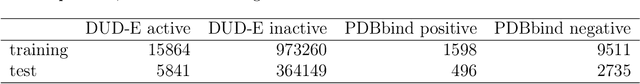
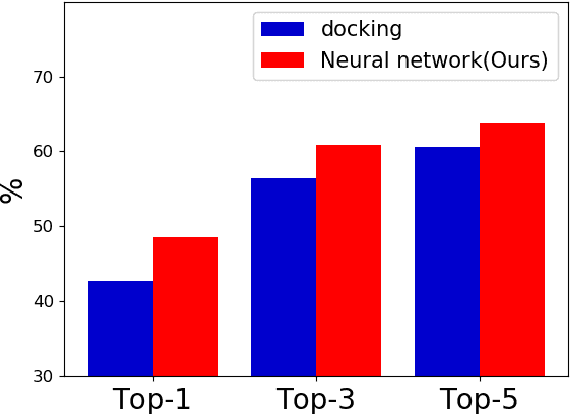

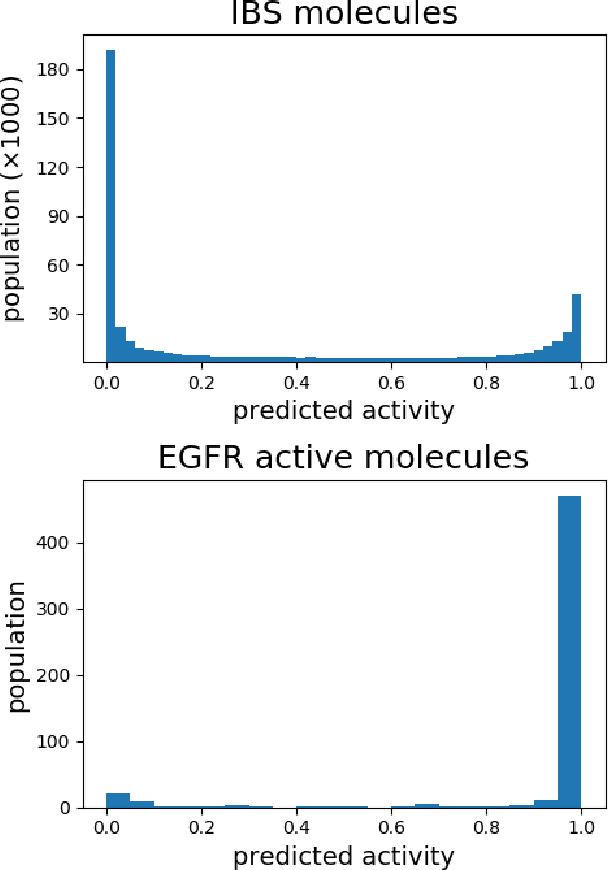
Accurate prediction of drug-target interaction (DTI) is essential for in silico drug design. For the purpose, we propose a novel approach for predicting DTI using a GNN that directly incorporates the 3D structure of a protein-ligand complex. We also apply a distance-aware graph attention algorithm with gate augmentation to increase the performance of our model. As a result, our model shows better performance than docking and other deep learning methods for both virtual screening and pose prediction. In addition, our model can reproduce the natural population distribution of active molecules and inactive molecules.
Uncertainty quantification of molecular property prediction with Bayesian neural networks
Mar 20, 2019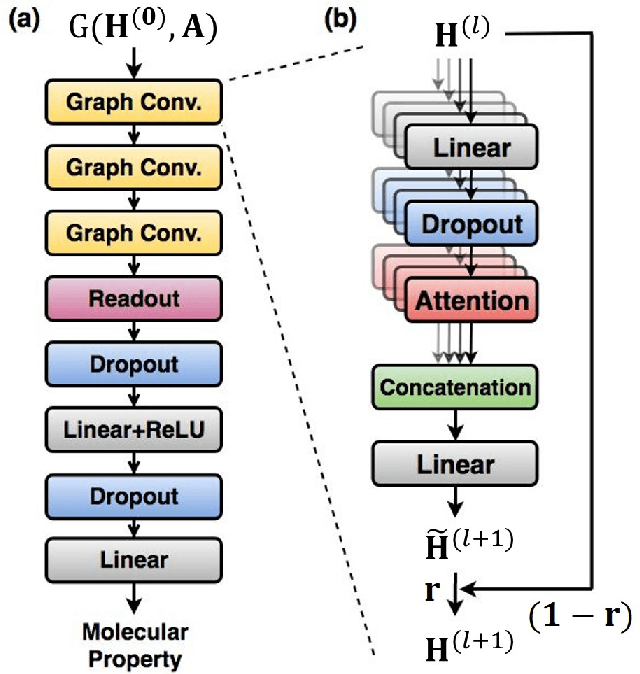


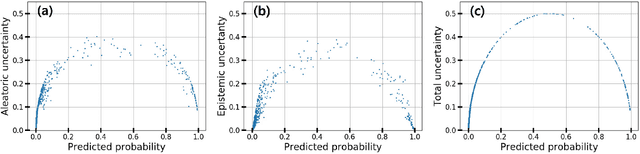
Deep neural networks have outperformed existing machine learning models in various molecular applications. In practical applications, it is still difficult to make confident decisions because of the uncertainty in predictions arisen from insufficient quality and quantity of training data. Here, we show that Bayesian neural networks are useful to quantify the uncertainty of molecular property prediction with three numerical experiments. In particular, it enables us to decompose the predictive variance into the model- and data-driven uncertainties, which helps to elucidate the source of errors. In the logP predictions, we show that data noise affected the data-driven uncertainties more significantly than the model-driven ones. Based on this analysis, we were able to find unexpected errors in the Harvard Clean Energy Project dataset. Lastly, we show that the confidence of prediction is closely related to the predictive uncertainty by performing on bio-activity and toxicity classification problems.
Deeply learning molecular structure-property relationships using attention- and gate-augmented graph convolutional network
Oct 08, 2018


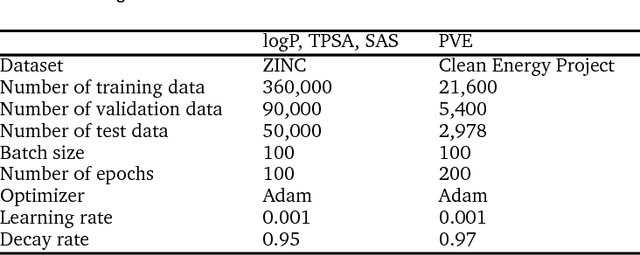
Molecular structure-property relationships are key to molecular engineering for materials and drug discovery. The rise of deep learning offers a new viable solution to elucidate the structure-property relationships directly from chemical data. Here we show that the performance of graph convolutional networks (GCNs) for the prediction of molecular properties can be improved by incorporating attention and gate mechanisms. The attention mechanism enables a GCN to identify atoms in different environments. The gated skip-connection further improves the GCN by updating feature maps at an appropriate rate. We demonstrate that the resulting attention- and gate-augmented GCN could extract better structural features related to a target molecular property such as solubility, polarity, synthetic accessibility and photovoltaic efficiency compared to the vanilla GCN. More interestingly, it identified two distinct parts of molecules as essential structural features for high photovoltaic efficiency, and each of them coincided with the areas of donor and acceptor orbitals for charge-transfer excitations, respectively. As a result, the new model could accurately predict molecular properties and place molecules with similar properties close to each other in a well-trained latent space, which is critical for successful molecular engineering.
Molecular generative model based on conditional variational autoencoder for de novo molecular design
Jun 15, 2018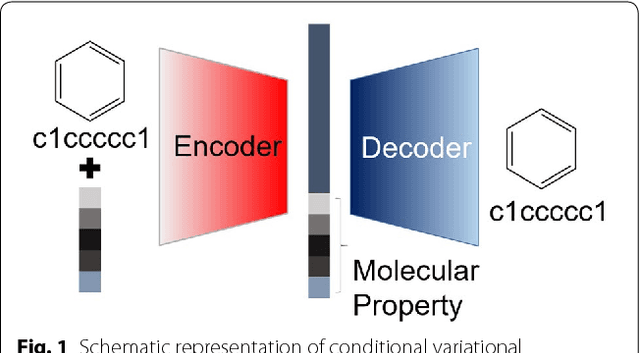


We propose a molecular generative model based on the conditional variational autoencoder for de novo molecular design. It is specialized to control multiple molecular properties simultaneously by imposing them on a latent space. As a proof of concept, we demonstrate that it can be used to generate drug-like molecules with five target properties. We were also able to adjust a single property without changing the others and to manipulate it beyond the range of the dataset.