 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProgressive Multi-Agent Reasoning for Biological Perturbation Prediction
Feb 07, 2026Predicting gene regulation responses to biological perturbations requires reasoning about underlying biological causalities. While large language models (LLMs) show promise for such tasks, they are often overwhelmed by the entangled nature of high-dimensional perturbation results. Moreover, recent works have primarily focused on genetic perturbations in single-cell experiments, leaving bulk-cell chemical perturbations, which is central to drug discovery, largely unexplored. Motivated by this, we present LINCSQA, a novel benchmark for predicting target gene regulation under complex chemical perturbations in bulk-cell environments. We further propose PBio-Agent, a multi-agent framework that integrates difficulty-aware task sequencing with iterative knowledge refinement. Our key insight is that genes affected by the same perturbation share causal structure, allowing confidently predicted genes to contextualize more challenging cases. The framework employs specialized agents enriched with biological knowledge graphs, while a synthesis agent integrates outputs and specialized judges ensure logical coherence. PBio-Agent outperforms existing baselines on both LINCSQA and PerturbQA, enabling even smaller models to predict and explain complex biological processes without additional training.
Compositional Flows for 3D Molecule and Synthesis Pathway Co-design
Apr 10, 2025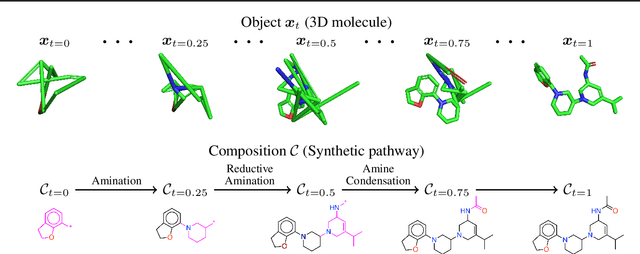
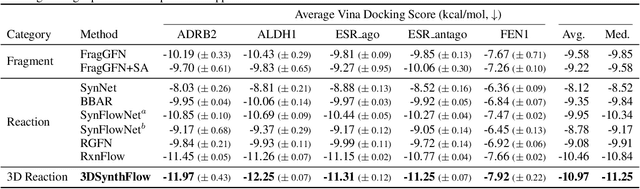
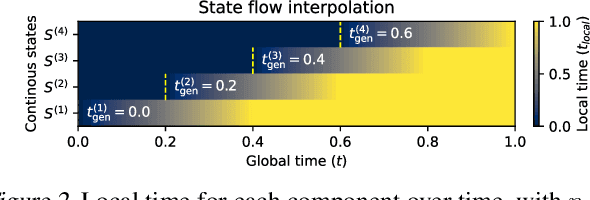
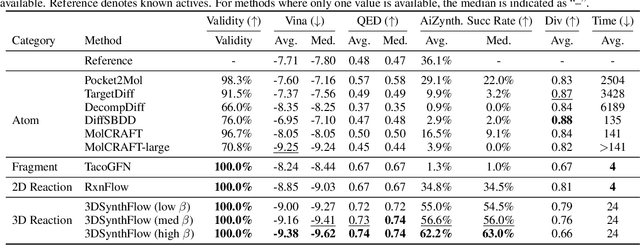
Many generative applications, such as synthesis-based 3D molecular design, involve constructing compositional objects with continuous features. Here, we introduce Compositional Generative Flows (CGFlow), a novel framework that extends flow matching to generate objects in compositional steps while modeling continuous states. Our key insight is that modeling compositional state transitions can be formulated as a straightforward extension of the flow matching interpolation process. We further build upon the theoretical foundations of generative flow networks (GFlowNets), enabling reward-guided sampling of compositional structures. We apply CGFlow to synthesizable drug design by jointly designing the molecule's synthetic pathway with its 3D binding pose. Our approach achieves state-of-the-art binding affinity on all 15 targets from the LIT-PCBA benchmark, and 5.8$\times$ improvement in sampling efficiency compared to 2D synthesis-based baseline. To our best knowledge, our method is also the first to achieve state of-art-performance in both Vina Dock (-9.38) and AiZynth success rate (62.2\%) on the CrossDocked benchmark.
Riemannian Denoising Score Matching for Molecular Structure Optimization with Accurate Energy
Nov 29, 2024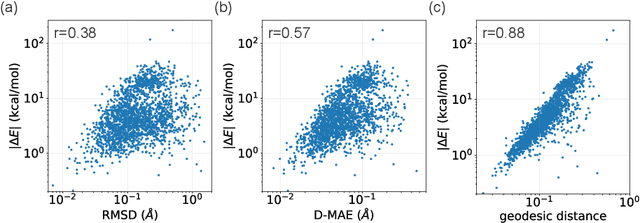
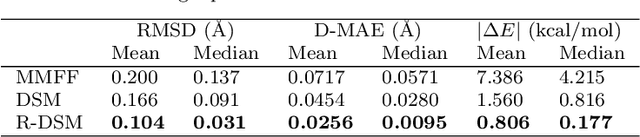
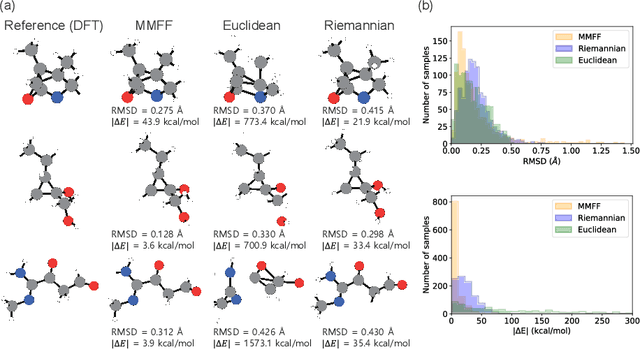

This study introduces a modified score matching method aimed at generating molecular structures with high energy accuracy. The denoising process of score matching or diffusion models mirrors molecular structure optimization, where scores act like physical force fields that guide particles toward equilibrium states. To achieve energetically accurate structures, it can be advantageous to have the score closely approximate the gradient of the actual potential energy surface. Unlike conventional methods that simply design the target score based on structural differences in Euclidean space, we propose a Riemannian score matching approach. This method represents molecular structures on a manifold defined by physics-informed internal coordinates to efficiently mimic the energy landscape, and performs noising and denoising within this space. Our method has been evaluated by refining several types of starting structures on the QM9 and GEOM datasets, demonstrating that the proposed Riemannian score matching method significantly improves the accuracy of the generated molecular structures, attaining chemical accuracy. The implications of this study extend to various applications in computational chemistry, offering a robust tool for accurate molecular structure prediction.
Generative Flows on Synthetic Pathway for Drug Design
Oct 06, 2024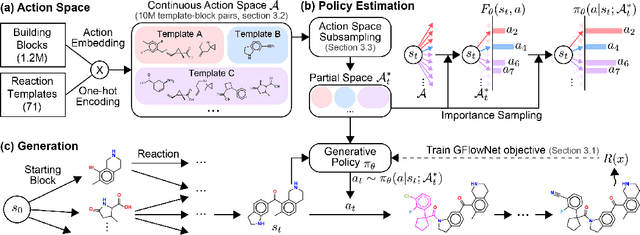
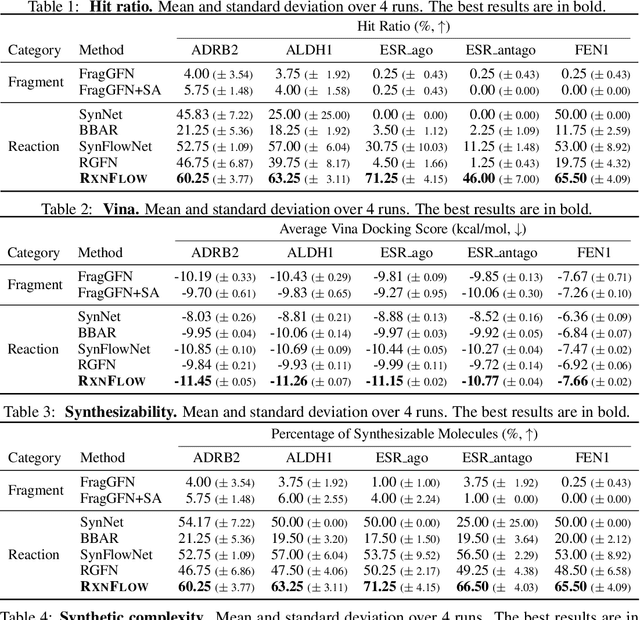
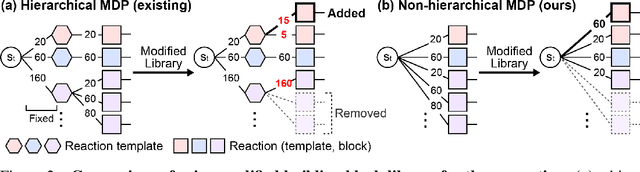
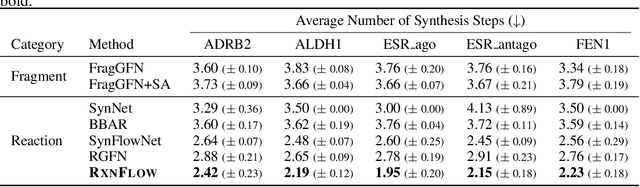
Generative models in drug discovery have recently gained attention as efficient alternatives to brute-force virtual screening. However, most existing models do not account for synthesizability, limiting their practical use in real-world scenarios. In this paper, we propose RxnFlow, which sequentially assembles molecules using predefined molecular building blocks and chemical reaction templates to constrain the synthetic chemical pathway. We then train on this sequential generating process with the objective of generative flow networks (GFlowNets) to generate both highly rewarded and diverse molecules. To mitigate the large action space of synthetic pathways in GFlowNets, we implement a novel action space subsampling method. This enables RxnFlow to learn generative flows over extensive action spaces comprising combinations of 1.2 million building blocks and 71 reaction templates without significant computational overhead. Additionally, RxnFlow can employ modified or expanded action spaces for generation without retraining, allowing for the introduction of additional objectives or the incorporation of newly discovered building blocks. We experimentally demonstrate that RxnFlow outperforms existing reaction-based and fragment-based models in pocket-specific optimization across various target pockets. Furthermore, RxnFlow achieves state-of-the-art performance on CrossDocked2020 for pocket-conditional generation, with an average Vina score of -8.85kcal/mol and 34.8% synthesizability.
Discrete Diffusion Schrödinger Bridge Matching for Graph Transformation
Oct 02, 2024



Transporting between arbitrary distributions is a fundamental goal in generative modeling. Recently proposed diffusion bridge models provide a potential solution, but they rely on a joint distribution that is difficult to obtain in practice. Furthermore, formulations based on continuous domains limit their applicability to discrete domains such as graphs. To overcome these limitations, we propose Discrete Diffusion Schr\"odinger Bridge Matching (DDSBM), a novel framework that utilizes continuous-time Markov chains to solve the SB problem in a high-dimensional discrete state space. Our approach extends Iterative Markovian Fitting to discrete domains, and we have proved its convergence to the SB. Furthermore, we adapt our framework for the graph transformation and show that our design choice of underlying dynamics characterized by independent modifications of nodes and edges can be interpreted as the entropy-regularized version of optimal transport with a cost function described by the graph edit distance. To demonstrate the effectiveness of our framework, we have applied DDSBM to molecular optimization in the field of chemistry. Experimental results demonstrate that DDSBM effectively optimizes molecules' property-of-interest with minimal graph transformation, successfully retaining other features.
Collective Variable Free Transition Path Sampling with Generative Flow Network
May 31, 2024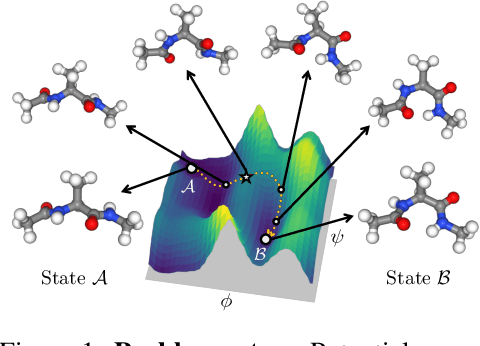
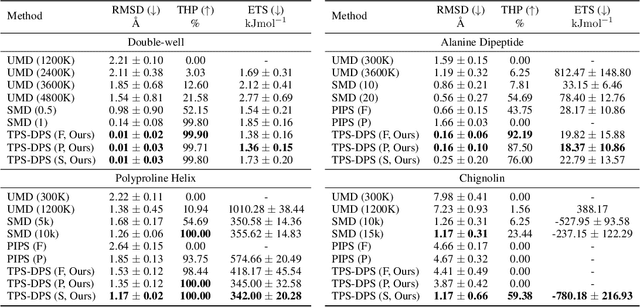
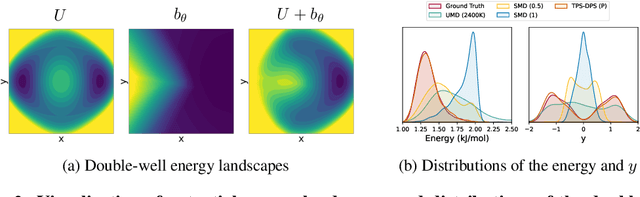

Understanding transition paths between meta-stable states in molecular systems is fundamental for material design and drug discovery. However, sampling these paths via molecular dynamics simulations is computationally prohibitive due to the high-energy barriers between the meta-stable states. Recent machine learning approaches are often restricted to simple systems or rely on collective variables (CVs) extracted from expensive domain knowledge. In this work, we propose to leverage generative flow networks (GFlowNets) to sample transition paths without relying on CVs. We reformulate the problem as amortized energy-based sampling over molecular trajectories and train a bias potential by minimizing the squared log-ratio between the target distribution and the generator, derived from the flow matching objective of GFlowNets. Our evaluation on three proteins (Alanine Dipeptide, Polyproline, and Chignolin) demonstrates that our approach, called TPS-GFN, generates more realistic and diverse transition paths than the previous CV-free machine learning approach.
NCIDiff: Non-covalent Interaction-generative Diffusion Model for Improving Reliability of 3D Molecule Generation Inside Protein Pocket
May 27, 2024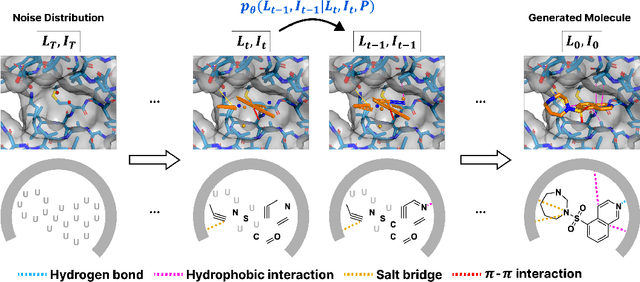
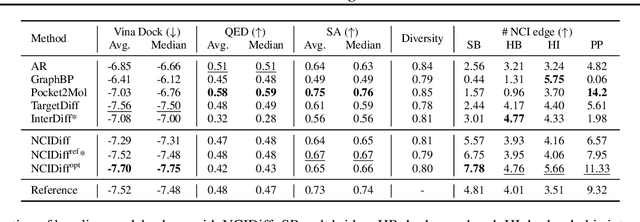
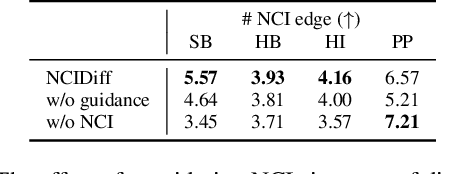
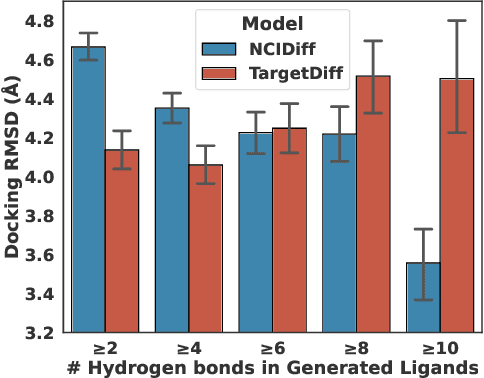
Advancements in deep generative modeling have changed the paradigm of drug discovery. Among such approaches, target-aware methods that exploit 3D structures of protein pockets were spotlighted for generating ligand molecules with their plausible binding modes. While docking scores superficially assess the quality of generated ligands, closer inspection of the binding structures reveals the inconsistency in local interactions between a pocket and generated ligands. Here, we address the issue by explicitly generating non-covalent interactions (NCIs), which are universal patterns throughout protein-ligand complexes. Our proposed model, NCIDiff, simultaneously denoises NCI types of protein-ligand edges along with a 3D graph of a ligand molecule during the sampling. With the NCI-generating strategy, our model generates ligands with more reliable NCIs, especially outperforming the baseline diffusion-based models. We further adopted inpainting techniques on NCIs to further improve the quality of the generated molecules. Finally, we showcase the applicability of NCIDiff on drug design tasks for real-world settings with specialized objectives by guiding the generation process with desired NCI patterns.
PharmacoNet: Accelerating Large-Scale Virtual Screening by Deep Pharmacophore Modeling
Oct 04, 2023



As the size of accessible compound libraries expands to over 10 billion, the need for more efficient structure-based virtual screening methods is emerging. Different pre-screening methods have been developed to rapidly screen the library, but the structure-based methods applicable to general proteins are still lacking: the challenge is to predict the binding pose between proteins and ligands and perform scoring in an extremely short time. We introduce PharmacoNet, a deep learning framework that identifies the optimal 3D pharmacophore arrangement which a ligand should have for stable binding from the binding site. By coarse-grained graph matching between ligands and the generated pharmacophore arrangement, we solve the expensive binding pose sampling and scoring procedures of existing methods in a single step. PharmacoNet is significantly faster than state-of-the-art structure-based approaches, yet reasonably accurate with a simple scoring function. Furthermore, we show the promising result that PharmacoNet effectively retains hit candidates even under the high pre-screening filtration rates. Overall, our study uncovers the hitherto untapped potential of a pharmacophore modeling approach in deep learning-based drug discovery.
C3Net: interatomic potential neural network for prediction of physicochemical properties in heterogenous systems
Sep 27, 2023



Understanding the interactions of a solute with its environment is of fundamental importance in chemistry and biology. In this work, we propose a deep neural network architecture for atom type embeddings in its molecular context and interatomic potential that follows fundamental physical laws. The architecture is applied to predict physicochemical properties in heterogeneous systems including solvation in diverse solvents, 1-octanol-water partitioning, and PAMPA with a single set of network weights. We show that our architecture is generalized well to the physicochemical properties and outperforms state-of-the-art approaches based on quantum mechanics and neural networks in the task of solvation free energy prediction. The interatomic potentials at each atom in a solute obtained from the model allow quantitative analysis of the physicochemical properties at atomic resolution consistent with chemical and physical reasoning. The software is available at https://github.com/SehanLee/C3Net.
PIGNet2: A Versatile Deep Learning-based Protein-Ligand Interaction Prediction Model for Binding Affinity Scoring and Virtual Screening
Jul 17, 2023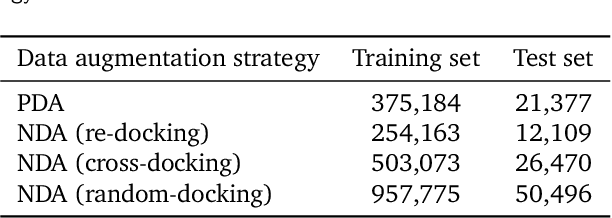
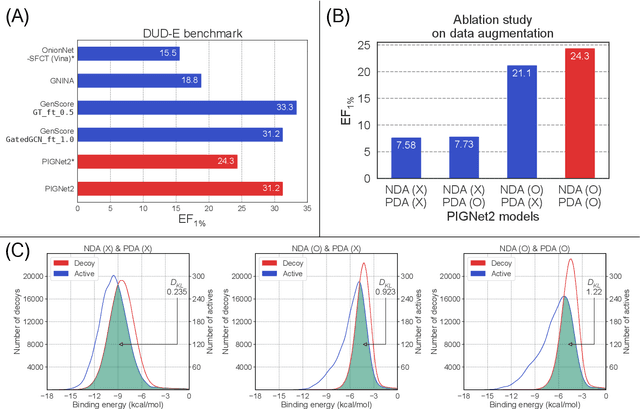
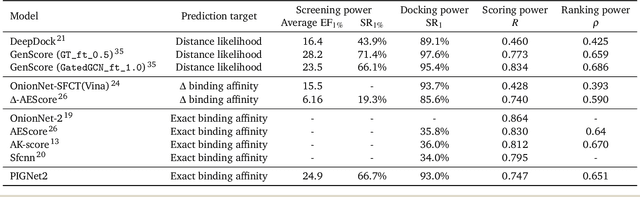
Prediction of protein-ligand interactions (PLI) plays a crucial role in drug discovery as it guides the identification and optimization of molecules that effectively bind to target proteins. Despite remarkable advances in deep learning-based PLI prediction, the development of a versatile model capable of accurately scoring binding affinity and conducting efficient virtual screening remains a challenge. The main obstacle in achieving this lies in the scarcity of experimental structure-affinity data, which limits the generalization ability of existing models. Here, we propose a viable solution to address this challenge by introducing a novel data augmentation strategy combined with a physics-informed graph neural network. The model showed significant improvements in both scoring and screening, outperforming task-specific deep learning models in various tests including derivative benchmarks, and notably achieving results comparable to the state-of-the-art performance based on distance likelihood learning. This demonstrates the potential of this approach to drug discovery.