 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePIGNet2: A Versatile Deep Learning-based Protein-Ligand Interaction Prediction Model for Binding Affinity Scoring and Virtual Screening
Paper and Code
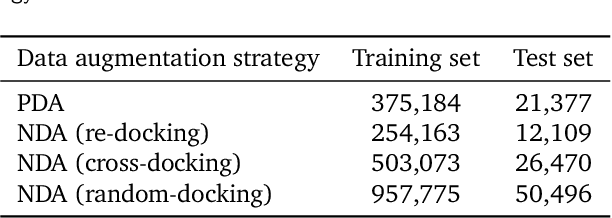
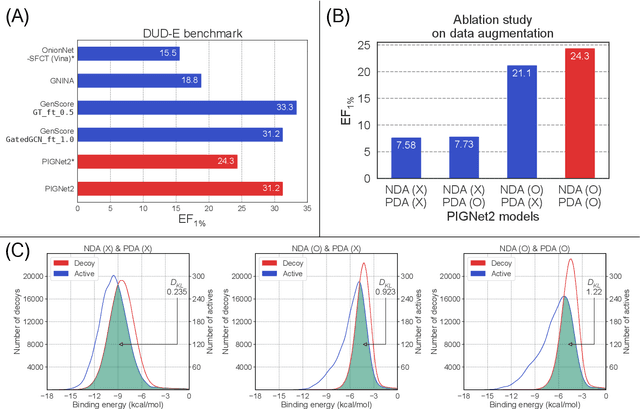
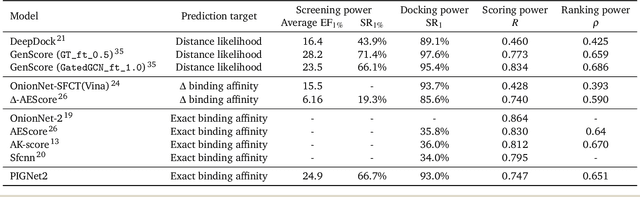
Prediction of protein-ligand interactions (PLI) plays a crucial role in drug discovery as it guides the identification and optimization of molecules that effectively bind to target proteins. Despite remarkable advances in deep learning-based PLI prediction, the development of a versatile model capable of accurately scoring binding affinity and conducting efficient virtual screening remains a challenge. The main obstacle in achieving this lies in the scarcity of experimental structure-affinity data, which limits the generalization ability of existing models. Here, we propose a viable solution to address this challenge by introducing a novel data augmentation strategy combined with a physics-informed graph neural network. The model showed significant improvements in both scoring and screening, outperforming task-specific deep learning models in various tests including derivative benchmarks, and notably achieving results comparable to the state-of-the-art performance based on distance likelihood learning. This demonstrates the potential of this approach to drug discovery.