 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDiscrete Diffusion Schrödinger Bridge Matching for Graph Transformation
Oct 02, 2024



Transporting between arbitrary distributions is a fundamental goal in generative modeling. Recently proposed diffusion bridge models provide a potential solution, but they rely on a joint distribution that is difficult to obtain in practice. Furthermore, formulations based on continuous domains limit their applicability to discrete domains such as graphs. To overcome these limitations, we propose Discrete Diffusion Schr\"odinger Bridge Matching (DDSBM), a novel framework that utilizes continuous-time Markov chains to solve the SB problem in a high-dimensional discrete state space. Our approach extends Iterative Markovian Fitting to discrete domains, and we have proved its convergence to the SB. Furthermore, we adapt our framework for the graph transformation and show that our design choice of underlying dynamics characterized by independent modifications of nodes and edges can be interpreted as the entropy-regularized version of optimal transport with a cost function described by the graph edit distance. To demonstrate the effectiveness of our framework, we have applied DDSBM to molecular optimization in the field of chemistry. Experimental results demonstrate that DDSBM effectively optimizes molecules' property-of-interest with minimal graph transformation, successfully retaining other features.
PIGNet2: A Versatile Deep Learning-based Protein-Ligand Interaction Prediction Model for Binding Affinity Scoring and Virtual Screening
Jul 17, 2023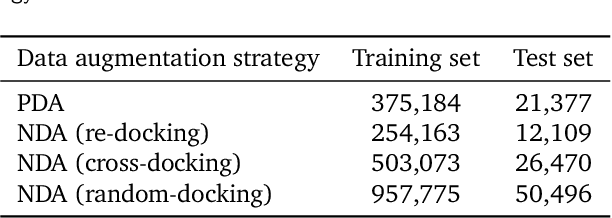
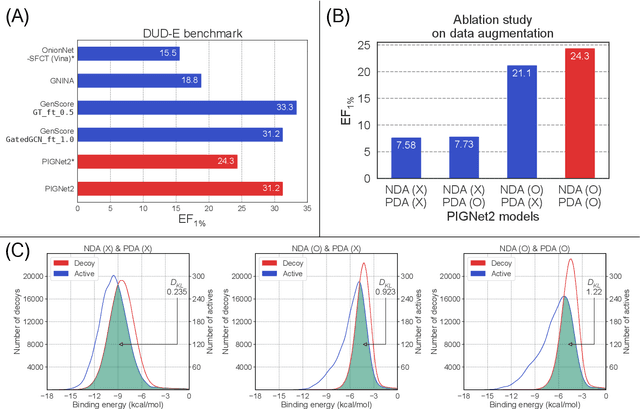
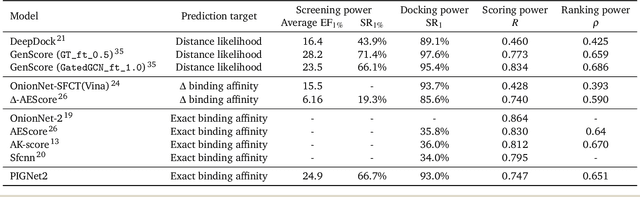
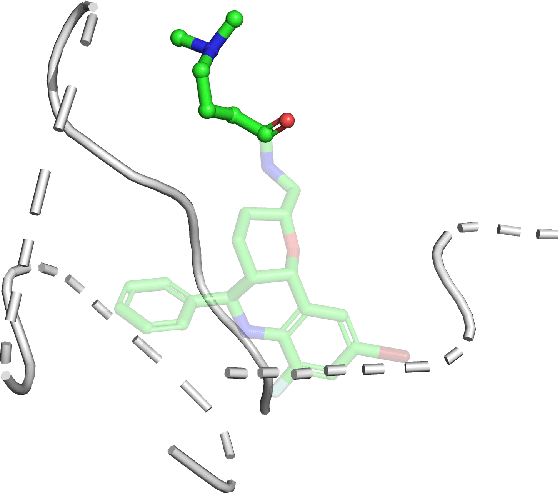
Prediction of protein-ligand interactions (PLI) plays a crucial role in drug discovery as it guides the identification and optimization of molecules that effectively bind to target proteins. Despite remarkable advances in deep learning-based PLI prediction, the development of a versatile model capable of accurately scoring binding affinity and conducting efficient virtual screening remains a challenge. The main obstacle in achieving this lies in the scarcity of experimental structure-affinity data, which limits the generalization ability of existing models. Here, we propose a viable solution to address this challenge by introducing a novel data augmentation strategy combined with a physics-informed graph neural network. The model showed significant improvements in both scoring and screening, outperforming task-specific deep learning models in various tests including derivative benchmarks, and notably achieving results comparable to the state-of-the-art performance based on distance likelihood learning. This demonstrates the potential of this approach to drug discovery.
Predicting quantum chemical property with easy-to-obtain geometry via positional denoising
Mar 28, 2023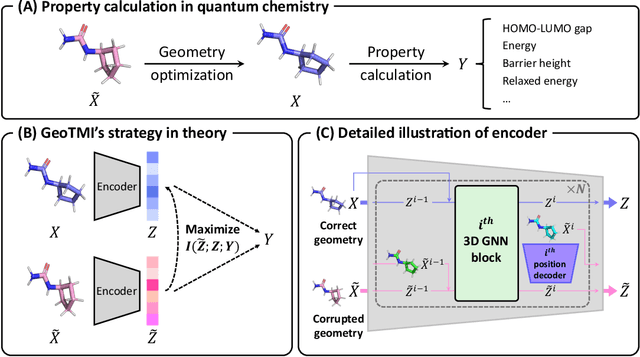
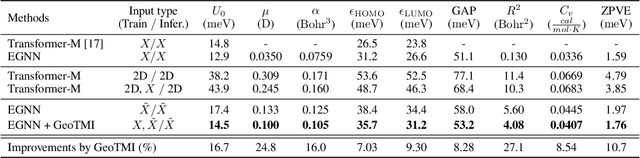
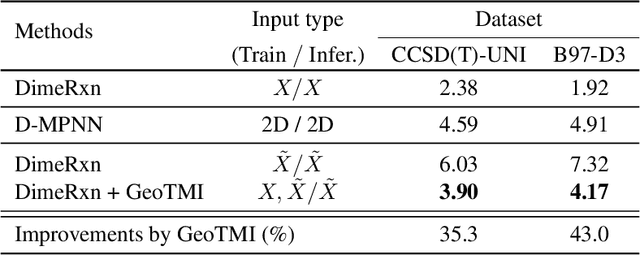
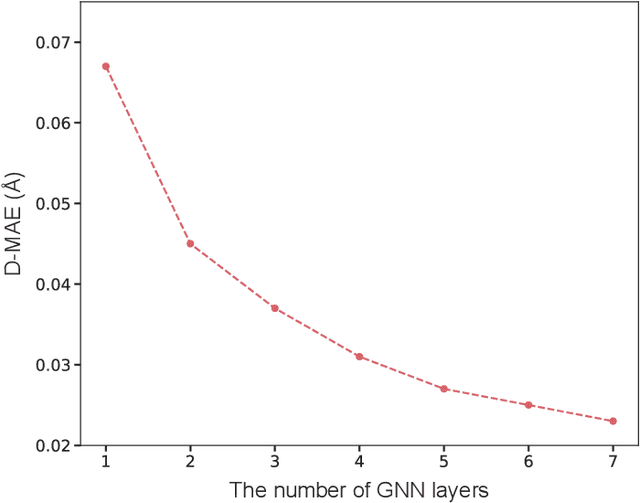
As quantum chemical properties have a significant dependence on their geometries, graph neural networks (GNNs) using 3D geometric information have achieved high prediction accuracy in many tasks. However, they often require 3D geometries obtained from high-level quantum mechanical calculations, which are practically infeasible, limiting their applicability in real-world problems. To tackle this, we propose a method to accurately predict the properties with relatively easy-to-obtain geometries (e.g., optimized geometries from the molecular force field). In this method, the input geometry, regarded as the corrupted geometry of the correct one, gradually approaches the correct one as it passes through the stacked denoising layers. We investigated the performance of the proposed method using 3D message-passing architectures for two prediction tasks: molecular properties and chemical reaction property. The reduction of positional errors through the denoising process contributed to performance improvement by increasing the mutual information between the correct and corrupted geometries. Moreover, our analysis of the correlation between denoising power and predictive accuracy demonstrates the effectiveness of the denoising process.
PIGNet: A physics-informed deep learning model toward generalized drug-target interaction predictions
Aug 22, 2020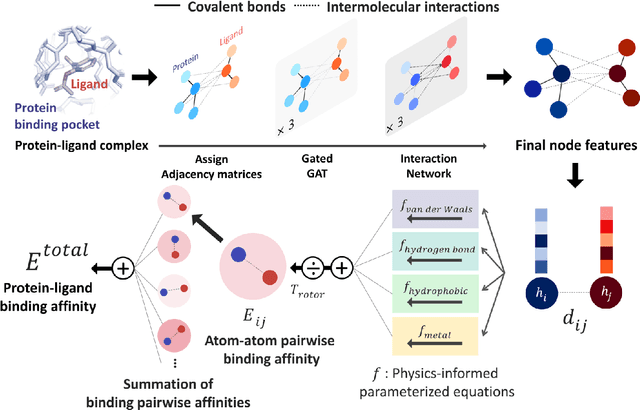
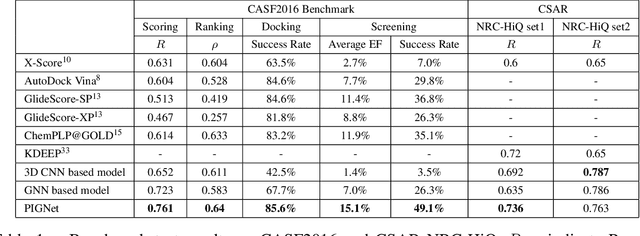
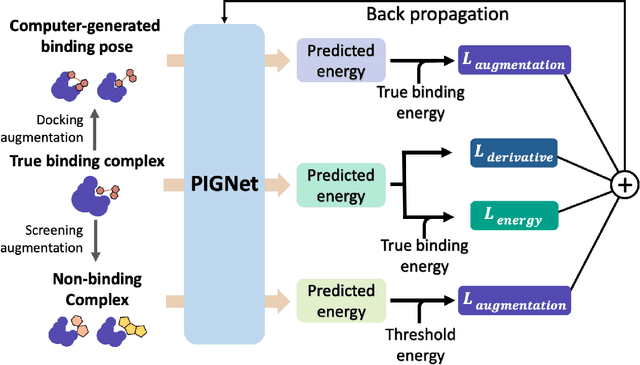
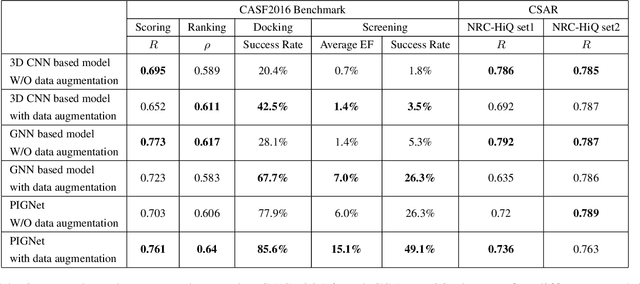
Recently, deep neural network (DNN)-based drug-target interaction (DTI) models are highlighted for their high accuracy with affordable computational costs. Yet, the models' insufficient generalization remains a challenging problem in the practice of in-silico drug discovery. We propose two key strategies to enhance generalization in the DTI model. The first one is to integrate physical models into DNN models. Our model, PIGNet, predicts the atom-atom pairwise interactions via physics-informed equations parameterized with neural networks and provides the total binding affinity of a protein-ligand complex as their sum. We further improved the model generalization by augmenting a wider range of binding poses and ligands to training data. PIGNet achieved a significant improvement in docking success rate, screening enhancement factor, and screening success rate by up to 2.01, 10.78, 14.0 times, respectively, compared to the previous DNN models. The physics-informed model also enables the interpretation of predicted binding affinities by visualizing the energy contribution of ligand substructures, providing insights for ligand optimization. Finally, we devised the uncertainty estimator of our model's prediction to qualify the outcomes and reduce the false positive rates.
Scaffold-based molecular design using graph generative model
May 31, 2019
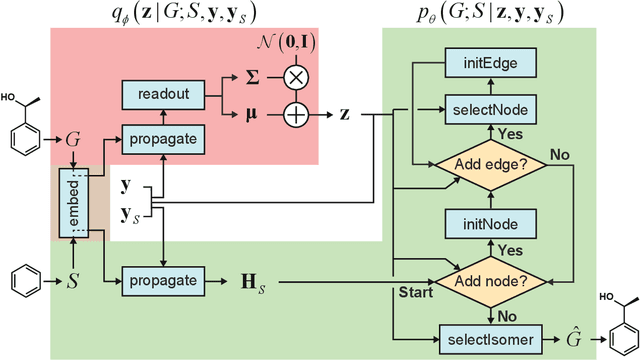
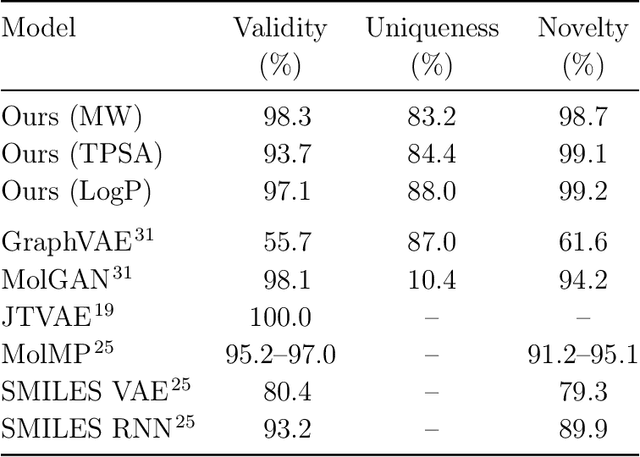
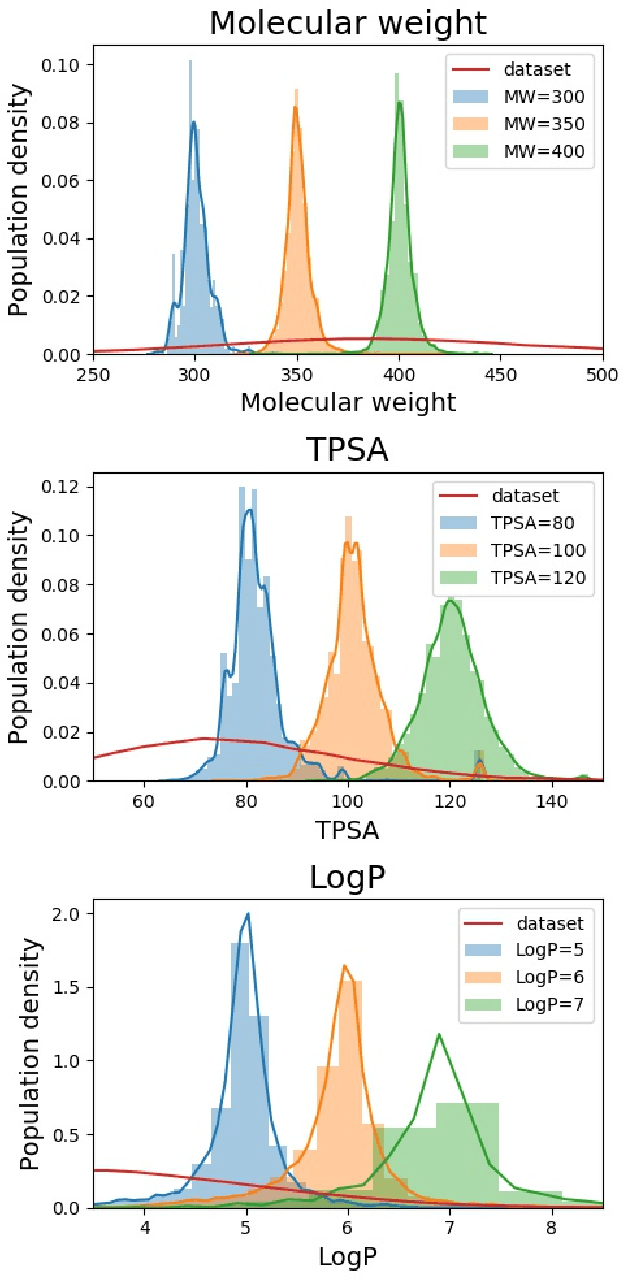
Searching new molecules in areas like drug discovery often starts from the core structures of candidate molecules to optimize the properties of interest. The way as such has called for a strategy of designing molecules retaining a particular scaffold as a substructure. On this account, our present work proposes a scaffold-based molecular generative model. The model generates molecular graphs by extending the graph of a scaffold through sequential additions of vertices and edges. In contrast to previous related models, our model guarantees the generated molecules to retain the given scaffold with certainty. Our evaluation of the model using unseen scaffolds showed the validity, uniqueness, and novelty of generated molecules as high as the case using seen scaffolds. This confirms that the model can generalize the learned chemical rules of adding atoms and bonds rather than simply memorizing the mapping from scaffolds to molecules during learning. Furthermore, despite the restraint of fixing core structures, our model could simultaneously control multiple molecular properties when generating new molecules.