 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEXAONE 4.5 Technical Report
Apr 09, 2026This technical report introduces EXAONE 4.5, the first open-weight vision language model released by LG AI Research. EXAONE 4.5 is architected by integrating a dedicated visual encoder into the existing EXAONE 4.0 framework, enabling native multimodal pretraining over both visual and textual modalities. The model is trained on large-scale data with careful curation, particularly emphasizing document-centric corpora that align with LG's strategic application domains. This targeted data design enables substantial performance gains in document understanding and related tasks, while also delivering broad improvements across general language capabilities. EXAONE 4.5 extends context length up to 256K tokens, facilitating long-context reasoning and enterprise-scale use cases. Comparative evaluations demonstrate that EXAONE 4.5 achieves competitive performance in general benchmarks while outperforming state-of-the-art models of similar scale in document understanding and Korean contextual reasoning. As part of LG's ongoing effort toward practical industrial deployment, EXAONE 4.5 is designed to be continuously extended with additional domains and application scenarios to advance AI for a better life.
K-EXAONE Technical Report
Jan 05, 2026This technical report presents K-EXAONE, a large-scale multilingual language model developed by LG AI Research. K-EXAONE is built on a Mixture-of-Experts architecture with 236B total parameters, activating 23B parameters during inference. It supports a 256K-token context window and covers six languages: Korean, English, Spanish, German, Japanese, and Vietnamese. We evaluate K-EXAONE on a comprehensive benchmark suite spanning reasoning, agentic, general, Korean, and multilingual abilities. Across these evaluations, K-EXAONE demonstrates performance comparable to open-weight models of similar size. K-EXAONE, designed to advance AI for a better life, is positioned as a powerful proprietary AI foundation model for a wide range of industrial and research applications.
EXAONE 4.0: Unified Large Language Models Integrating Non-reasoning and Reasoning Modes
Jul 15, 2025This technical report introduces EXAONE 4.0, which integrates a Non-reasoning mode and a Reasoning mode to achieve both the excellent usability of EXAONE 3.5 and the advanced reasoning abilities of EXAONE Deep. To pave the way for the agentic AI era, EXAONE 4.0 incorporates essential features such as agentic tool use, and its multilingual capabilities are extended to support Spanish in addition to English and Korean. The EXAONE 4.0 model series consists of two sizes: a mid-size 32B model optimized for high performance, and a small-size 1.2B model designed for on-device applications. The EXAONE 4.0 demonstrates superior performance compared to open-weight models in its class and remains competitive even against frontier-class models. The models are publicly available for research purposes and can be easily downloaded via https://huggingface.co/LGAI-EXAONE.
EXAONE Deep: Reasoning Enhanced Language Models
Mar 16, 2025We present EXAONE Deep series, which exhibits superior capabilities in various reasoning tasks, including math and coding benchmarks. We train our models mainly on the reasoning-specialized dataset that incorporates long streams of thought processes. Evaluation results show that our smaller models, EXAONE Deep 2.4B and 7.8B, outperform other models of comparable size, while the largest model, EXAONE Deep 32B, demonstrates competitive performance against leading open-weight models. All EXAONE Deep models are openly available for research purposes and can be downloaded from https://huggingface.co/LGAI-EXAONE
FragFM: Efficient Fragment-Based Molecular Generation via Discrete Flow Matching
Feb 19, 2025



We introduce FragFM, a novel fragment-based discrete flow matching framework for molecular graph generation.FragFM generates molecules at the fragment level, leveraging a coarse-to-fine autoencoding mechanism to reconstruct atom-level details. This approach reduces computational complexity while maintaining high chemical validity, enabling more efficient and scalable molecular generation. We benchmark FragFM against state-of-the-art diffusion- and flow-based models on standard molecular generation benchmarks and natural product datasets, demonstrating superior performance in validity, property control, and sampling efficiency. Notably, FragFM achieves over 99\% validity with significantly fewer sampling steps, improving scalability while preserving molecular diversity. These results highlight the potential of fragment-based generative modeling for large-scale, property-aware molecular design, paving the way for more efficient exploration of chemical space.
EXAONE 3.5: Series of Large Language Models for Real-world Use Cases
Dec 09, 2024



This technical report introduces the EXAONE 3.5 instruction-tuned language models, developed and released by LG AI Research. The EXAONE 3.5 language models are offered in three configurations: 32B, 7.8B, and 2.4B. These models feature several standout capabilities: 1) exceptional instruction following capabilities in real-world scenarios, achieving the highest scores across seven benchmarks, 2) outstanding long-context comprehension, attaining the top performance in four benchmarks, and 3) competitive results compared to state-of-the-art open models of similar sizes across nine general benchmarks. The EXAONE 3.5 language models are open to anyone for research purposes and can be downloaded from https://huggingface.co/LGAI-EXAONE. For commercial use, please reach out to the official contact point of LG AI Research: contact_us@lgresearch.ai.
Riemannian Denoising Score Matching for Molecular Structure Optimization with Accurate Energy
Nov 29, 2024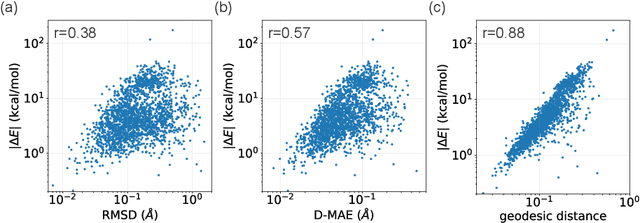
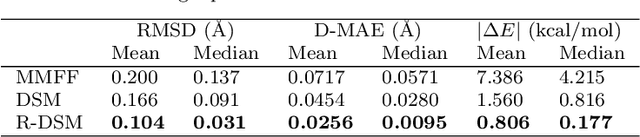
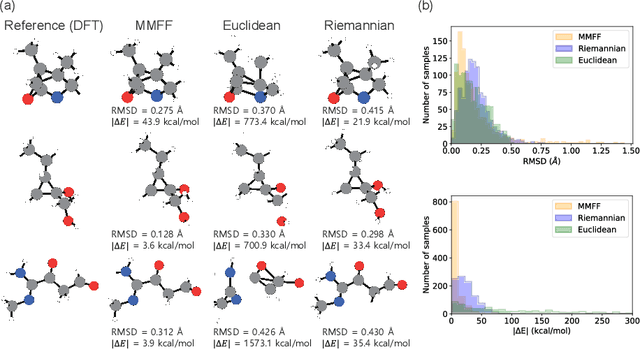

This study introduces a modified score matching method aimed at generating molecular structures with high energy accuracy. The denoising process of score matching or diffusion models mirrors molecular structure optimization, where scores act like physical force fields that guide particles toward equilibrium states. To achieve energetically accurate structures, it can be advantageous to have the score closely approximate the gradient of the actual potential energy surface. Unlike conventional methods that simply design the target score based on structural differences in Euclidean space, we propose a Riemannian score matching approach. This method represents molecular structures on a manifold defined by physics-informed internal coordinates to efficiently mimic the energy landscape, and performs noising and denoising within this space. Our method has been evaluated by refining several types of starting structures on the QM9 and GEOM datasets, demonstrating that the proposed Riemannian score matching method significantly improves the accuracy of the generated molecular structures, attaining chemical accuracy. The implications of this study extend to various applications in computational chemistry, offering a robust tool for accurate molecular structure prediction.
Discrete Diffusion Schrödinger Bridge Matching for Graph Transformation
Oct 02, 2024



Transporting between arbitrary distributions is a fundamental goal in generative modeling. Recently proposed diffusion bridge models provide a potential solution, but they rely on a joint distribution that is difficult to obtain in practice. Furthermore, formulations based on continuous domains limit their applicability to discrete domains such as graphs. To overcome these limitations, we propose Discrete Diffusion Schr\"odinger Bridge Matching (DDSBM), a novel framework that utilizes continuous-time Markov chains to solve the SB problem in a high-dimensional discrete state space. Our approach extends Iterative Markovian Fitting to discrete domains, and we have proved its convergence to the SB. Furthermore, we adapt our framework for the graph transformation and show that our design choice of underlying dynamics characterized by independent modifications of nodes and edges can be interpreted as the entropy-regularized version of optimal transport with a cost function described by the graph edit distance. To demonstrate the effectiveness of our framework, we have applied DDSBM to molecular optimization in the field of chemistry. Experimental results demonstrate that DDSBM effectively optimizes molecules' property-of-interest with minimal graph transformation, successfully retaining other features.
EXAONE 3.0 7.8B Instruction Tuned Language Model
Aug 07, 2024



We introduce EXAONE 3.0 instruction-tuned language model, the first open model in the family of Large Language Models (LLMs) developed by LG AI Research. Among different model sizes, we publicly release the 7.8B instruction-tuned model to promote open research and innovations. Through extensive evaluations across a wide range of public and in-house benchmarks, EXAONE 3.0 demonstrates highly competitive real-world performance with instruction-following capability against other state-of-the-art open models of similar size. Our comparative analysis shows that EXAONE 3.0 excels particularly in Korean, while achieving compelling performance across general tasks and complex reasoning. With its strong real-world effectiveness and bilingual proficiency, we hope that EXAONE keeps contributing to advancements in Expert AI. Our EXAONE 3.0 instruction-tuned model is available at https://huggingface.co/LGAI-EXAONE/EXAONE-3.0-7.8B-Instruct
Collective Variable Free Transition Path Sampling with Generative Flow Network
May 31, 2024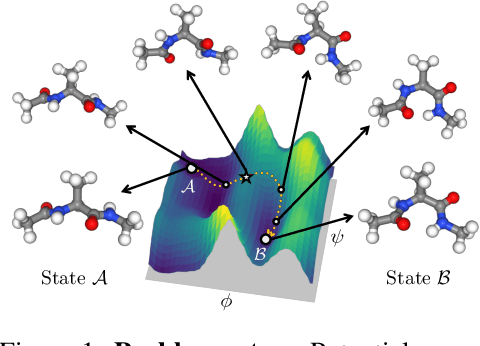
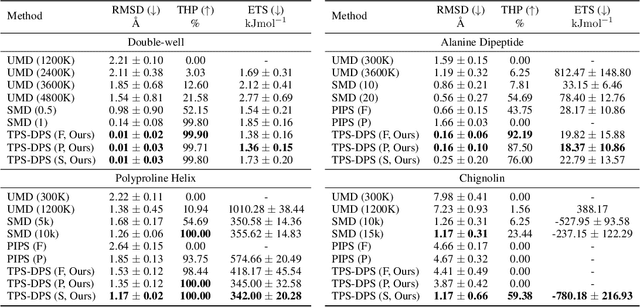
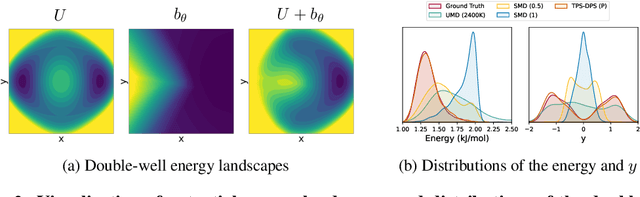

Understanding transition paths between meta-stable states in molecular systems is fundamental for material design and drug discovery. However, sampling these paths via molecular dynamics simulations is computationally prohibitive due to the high-energy barriers between the meta-stable states. Recent machine learning approaches are often restricted to simple systems or rely on collective variables (CVs) extracted from expensive domain knowledge. In this work, we propose to leverage generative flow networks (GFlowNets) to sample transition paths without relying on CVs. We reformulate the problem as amortized energy-based sampling over molecular trajectories and train a bias potential by minimizing the squared log-ratio between the target distribution and the generator, derived from the flow matching objective of GFlowNets. Our evaluation on three proteins (Alanine Dipeptide, Polyproline, and Chignolin) demonstrates that our approach, called TPS-GFN, generates more realistic and diverse transition paths than the previous CV-free machine learning approach.