 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRiemannian Denoising Score Matching for Molecular Structure Optimization with Accurate Energy
Paper and Code
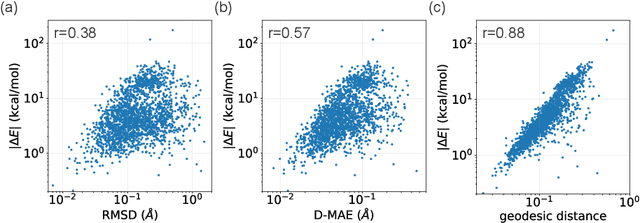
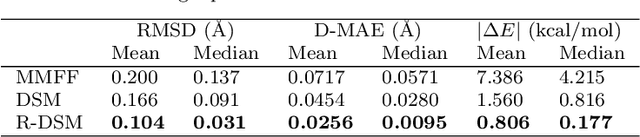
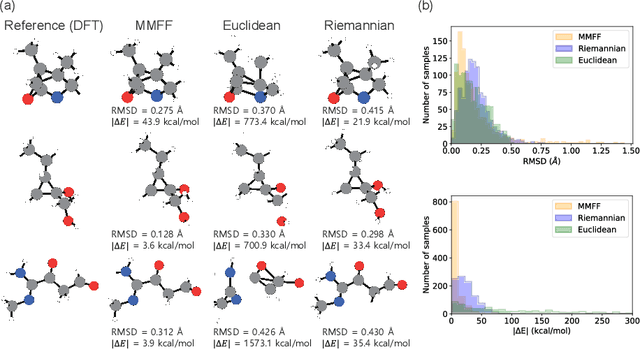

This study introduces a modified score matching method aimed at generating molecular structures with high energy accuracy. The denoising process of score matching or diffusion models mirrors molecular structure optimization, where scores act like physical force fields that guide particles toward equilibrium states. To achieve energetically accurate structures, it can be advantageous to have the score closely approximate the gradient of the actual potential energy surface. Unlike conventional methods that simply design the target score based on structural differences in Euclidean space, we propose a Riemannian score matching approach. This method represents molecular structures on a manifold defined by physics-informed internal coordinates to efficiently mimic the energy landscape, and performs noising and denoising within this space. Our method has been evaluated by refining several types of starting structures on the QM9 and GEOM datasets, demonstrating that the proposed Riemannian score matching method significantly improves the accuracy of the generated molecular structures, attaining chemical accuracy. The implications of this study extend to various applications in computational chemistry, offering a robust tool for accurate molecular structure prediction.