 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeINDIBATOR: Diverse and Fact-Grounded Individuality for Multi-Agent Debate in Molecular Discovery
Feb 02, 2026Multi-agent systems have emerged as a powerful paradigm for automating scientific discovery. To differentiate agent behavior in the multi-agent system, current frameworks typically assign generic role-based personas such as ''reviewer'' or ''writer'' or rely on coarse grained keyword-based personas. While functional, this approach oversimplifies how human scientists operate, whose contributions are shaped by their unique research trajectories. In response, we propose INDIBATOR, a framework for molecular discovery that grounds agents in individualized scientist profiles constructed from two modalities: publication history for literature-derived knowledge and molecular history for structural priors. These agents engage in multi-turn debate through proposal, critique, and voting phases. Our evaluation demonstrates that these fine-grained individuality-grounded agents consistently outperform systems relying on coarse-grained personas, achieving competitive or state-of-the-art performance. These results validate that capturing the ``scientific DNA'' of individual agents is essential for high-quality discovery.
DNACHUNKER: Learnable Tokenization for DNA Language Models
Jan 06, 2026DNA language models have emerged as powerful tools for decoding the complex language of DNA sequences. However, the performance of these models is heavily affected by their tokenization strategy, i.e., a method used to parse DNA sequences into a shorter sequence of chunks. In this work, we propose DNACHUNKER, which integrates a learnable dynamic DNA tokenization mechanism and is trained as a masked language model. Adopting the dynamic chunking procedure proposed by H-Net, our model learns to segment sequences into variable-length chunks. This dynamic chunking offers two key advantages: it's resilient to shifts and mutations in the DNA, and it allocates more detail to important functional areas. We demonstrate the performance of DNACHUNKER by training it on the human reference genome (HG38) and testing it on the Nucleotide Transformer and Genomic benchmarks. Further ablative experiments reveal that DNACHUNKER learns tokenization that grasps biological grammar and uses smaller chunks to preserve detail in important functional elements such as promoters and exons, while using larger chunks for repetitive, redundant regions.
Learning Collective Variables from Time-lagged Generation
Jul 10, 2025Rare events such as state transitions are difficult to observe directly with molecular dynamics simulations due to long timescales. Enhanced sampling techniques overcome this by introducing biases along carefully chosen low-dimensional features, known as collective variables (CVs), which capture the slow degrees of freedom. Machine learning approaches (MLCVs) have automated CV discovery, but existing methods typically focus on discriminating meta-stable states without fully encoding the detailed dynamics essential for accurate sampling. We propose TLC, a framework that learns CVs directly from time-lagged conditions of a generative model. Instead of modeling the static Boltzmann distribution, TLC models a time-lagged conditional distribution yielding CVs to capture the slow dynamic behavior. We validate TLC on the Alanine Dipeptide system using two CV-based enhanced sampling tasks: (i) steered molecular dynamics (SMD) and (ii) on-the-fly probability enhanced sampling (OPES), demonstrating equal or superior performance compared to existing MLCV methods in both transition path sampling and state discrimination.
Enhancing LLM Agent Safety via Causal Influence Prompting
Jul 01, 2025



As autonomous agents powered by large language models (LLMs) continue to demonstrate potential across various assistive tasks, ensuring their safe and reliable behavior is crucial for preventing unintended consequences. In this work, we introduce CIP, a novel technique that leverages causal influence diagrams (CIDs) to identify and mitigate risks arising from agent decision-making. CIDs provide a structured representation of cause-and-effect relationships, enabling agents to anticipate harmful outcomes and make safer decisions. Our approach consists of three key steps: (1) initializing a CID based on task specifications to outline the decision-making process, (2) guiding agent interactions with the environment using the CID, and (3) iteratively refining the CID based on observed behaviors and outcomes. Experimental results demonstrate that our method effectively enhances safety in both code execution and mobile device control tasks.
MT-Mol:Multi Agent System with Tool-based Reasoning for Molecular Optimization
May 27, 2025Large language models (LLMs) have large potential for molecular optimization, as they can gather external chemistry tools and enable collaborative interactions to iteratively refine molecular candidates. However, this potential remains underexplored, particularly in the context of structured reasoning, interpretability, and comprehensive tool-grounded molecular optimization. To address this gap, we introduce MT-Mol, a multi-agent framework for molecular optimization that leverages tool-guided reasoning and role-specialized LLM agents. Our system incorporates comprehensive RDKit tools, categorized into five distinct domains: structural descriptors, electronic and topological features, fragment-based functional groups, molecular representations, and miscellaneous chemical properties. Each category is managed by an expert analyst agent, responsible for extracting task-relevant tools and enabling interpretable, chemically grounded feedback. MT-Mol produces molecules with tool-aligned and stepwise reasoning through the interaction between the analyst agents, a molecule-generating scientist, a reasoning-output verifier, and a reviewer agent. As a result, we show that our framework shows the state-of-the-art performance of the PMO-1K benchmark on 17 out of 23 tasks.
Energy-based generator matching: A neural sampler for general state space
May 26, 2025
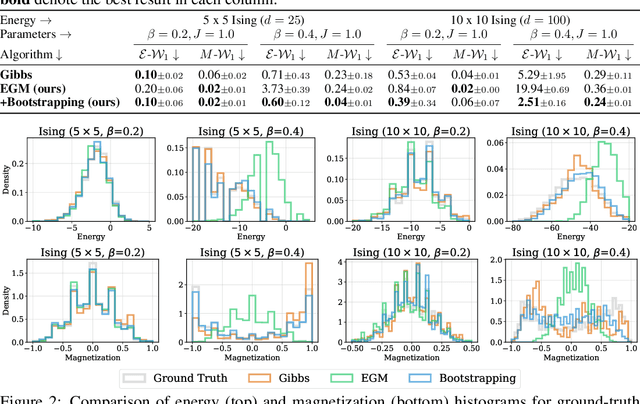
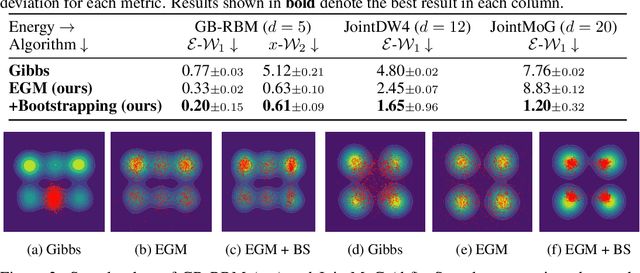
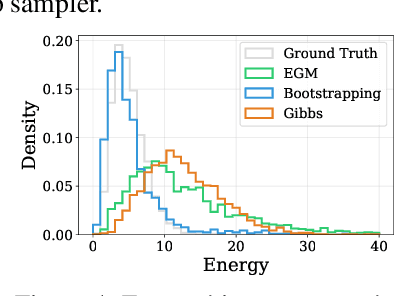
We propose Energy-based generator matching (EGM), a modality-agnostic approach to train generative models from energy functions in the absence of data. Extending the recently proposed generator matching, EGM enables training of arbitrary continuous-time Markov processes, e.g., diffusion, flow, and jump, and can generate data from continuous, discrete, and a mixture of two modalities. To this end, we propose estimating the generator matching loss using self-normalized importance sampling with an additional bootstrapping trick to reduce variance in the importance weight. We validate EGM on both discrete and multimodal tasks up to 100 and 20 dimensions, respectively.
On scalable and efficient training of diffusion samplers
May 26, 2025We address the challenge of training diffusion models to sample from unnormalized energy distributions in the absence of data, the so-called diffusion samplers. Although these approaches have shown promise, they struggle to scale in more demanding scenarios where energy evaluations are expensive and the sampling space is high-dimensional. To address this limitation, we propose a scalable and sample-efficient framework that properly harmonizes the powerful classical sampling method and the diffusion sampler. Specifically, we utilize Monte Carlo Markov chain (MCMC) samplers with a novelty-based auxiliary energy as a Searcher to collect off-policy samples, using an auxiliary energy function to compensate for exploring modes the diffusion sampler rarely visits. These off-policy samples are then combined with on-policy data to train the diffusion sampler, thereby expanding its coverage of the energy landscape. Furthermore, we identify primacy bias, i.e., the preference of samplers for early experience during training, as the main cause of mode collapse during training, and introduce a periodic re-initialization trick to resolve this issue. Our method significantly improves sample efficiency on standard benchmarks for diffusion samplers and also excels at higher-dimensional problems and real-world molecular conformer generation.
High-order Equivariant Flow Matching for Density Functional Theory Hamiltonian Prediction
May 24, 2025Density functional theory (DFT) is a fundamental method for simulating quantum chemical properties, but it remains expensive due to the iterative self-consistent field (SCF) process required to solve the Kohn-Sham equations. Recently, deep learning methods are gaining attention as a way to bypass this step by directly predicting the Hamiltonian. However, they rely on deterministic regression and do not consider the highly structured nature of Hamiltonians. In this work, we propose QHFlow, a high-order equivariant flow matching framework that generates Hamiltonian matrices conditioned on molecular geometry. Flow matching models continuous-time trajectories between simple priors and complex targets, learning the structured distributions over Hamiltonians instead of direct regression. To further incorporate symmetry, we use a neural architecture that predicts SE(3)-equivariant vector fields, improving accuracy and generalization across diverse geometries. To further enhance physical fidelity, we additionally introduce a fine-tuning scheme to align predicted orbital energies with the target. QHFlow achieves state-of-the-art performance, reducing Hamiltonian error by 71% on MD17 and 53% on QH9. Moreover, we further show that QHFlow accelerates the DFT process without trading off the solution quality when initializing SCF iterations with the predicted Hamiltonian, significantly reducing the number of iterations and runtime.
Self-Training Large Language Models with Confident Reasoning
May 23, 2025Large language models (LLMs) have shown impressive performance by generating reasoning paths before final answers, but learning such a reasoning path requires costly human supervision. To address this issue, recent studies have explored self-training methods that improve reasoning capabilities using pseudo-labels generated by the LLMs themselves. Among these, confidence-based self-training fine-tunes LLMs to prefer reasoning paths with high-confidence answers, where confidence is estimated via majority voting. However, such methods exclusively focus on the quality of the final answer and may ignore the quality of the reasoning paths, as even an incorrect reasoning path leads to a correct answer by chance. Instead, we advocate the use of reasoning-level confidence to identify high-quality reasoning paths for self-training, supported by our empirical observations. We then propose a new self-training method, CORE-PO, that fine-tunes LLMs to prefer high-COnfidence REasoning paths through Policy Optimization. Our experiments show that CORE-PO improves the accuracy of outputs on four in-distribution and two out-of-distribution benchmarks, compared to existing self-training methods.
Flexible MOF Generation with Torsion-Aware Flow Matching
May 23, 2025Designing metal-organic frameworks (MOFs) with novel chemistries is a long-standing challenge due to their large combinatorial space and the complex 3D arrangements of building blocks. While recent deep generative models have enabled scalable MOF generation, they assume (1) a fixed set of building blocks and (2) known ground-truth local block-wise 3D coordinates. However, this limits their ability to (1) design novel MOFs and (2) generate the structure using novel building blocks. We propose a two-stage de novo MOF generation framework that overcomes these limitations by modeling both chemical and geometric degrees of freedom. First, we train a SMILES-based autoregressive model to generate novel metal and organic building blocks, paired with cheminformatics for 3D structure initialization. Second, we introduce a flow-matching model that predicts translations, rotations, and torsional angles to assemble flexible blocks into valid 3D frameworks. Our experiments demonstrate improved reconstruction accuracy, the generation of valid, novel, and unique MOFs, and the ability of our model to create novel building blocks.