 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCollective Variable Free Transition Path Sampling with Generative Flow Network
Paper and Code
May 31, 2024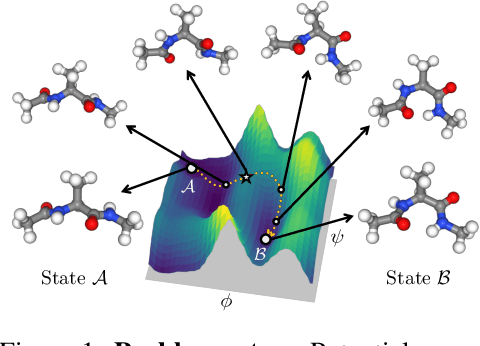
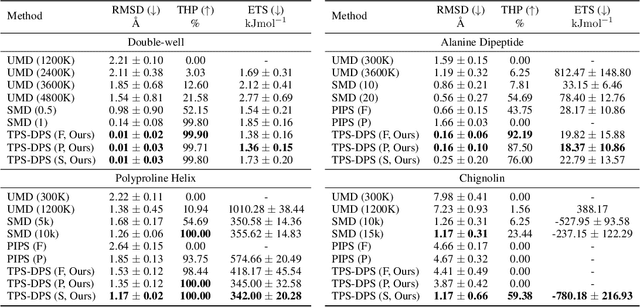
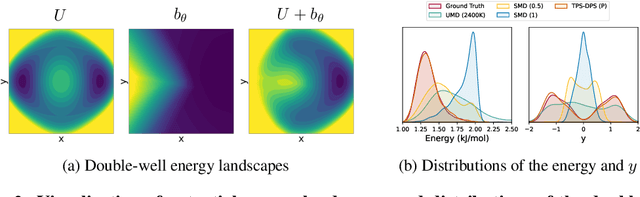

Understanding transition paths between meta-stable states in molecular systems is fundamental for material design and drug discovery. However, sampling these paths via molecular dynamics simulations is computationally prohibitive due to the high-energy barriers between the meta-stable states. Recent machine learning approaches are often restricted to simple systems or rely on collective variables (CVs) extracted from expensive domain knowledge. In this work, we propose to leverage generative flow networks (GFlowNets) to sample transition paths without relying on CVs. We reformulate the problem as amortized energy-based sampling over molecular trajectories and train a bias potential by minimizing the squared log-ratio between the target distribution and the generator, derived from the flow matching objective of GFlowNets. Our evaluation on three proteins (Alanine Dipeptide, Polyproline, and Chignolin) demonstrates that our approach, called TPS-GFN, generates more realistic and diverse transition paths than the previous CV-free machine learning approach.