 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProgressive Multi-Agent Reasoning for Biological Perturbation Prediction
Feb 07, 2026Predicting gene regulation responses to biological perturbations requires reasoning about underlying biological causalities. While large language models (LLMs) show promise for such tasks, they are often overwhelmed by the entangled nature of high-dimensional perturbation results. Moreover, recent works have primarily focused on genetic perturbations in single-cell experiments, leaving bulk-cell chemical perturbations, which is central to drug discovery, largely unexplored. Motivated by this, we present LINCSQA, a novel benchmark for predicting target gene regulation under complex chemical perturbations in bulk-cell environments. We further propose PBio-Agent, a multi-agent framework that integrates difficulty-aware task sequencing with iterative knowledge refinement. Our key insight is that genes affected by the same perturbation share causal structure, allowing confidently predicted genes to contextualize more challenging cases. The framework employs specialized agents enriched with biological knowledge graphs, while a synthesis agent integrates outputs and specialized judges ensure logical coherence. PBio-Agent outperforms existing baselines on both LINCSQA and PerturbQA, enabling even smaller models to predict and explain complex biological processes without additional training.
PIGNet2: A Versatile Deep Learning-based Protein-Ligand Interaction Prediction Model for Binding Affinity Scoring and Virtual Screening
Jul 17, 2023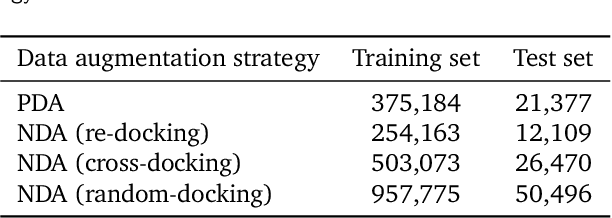
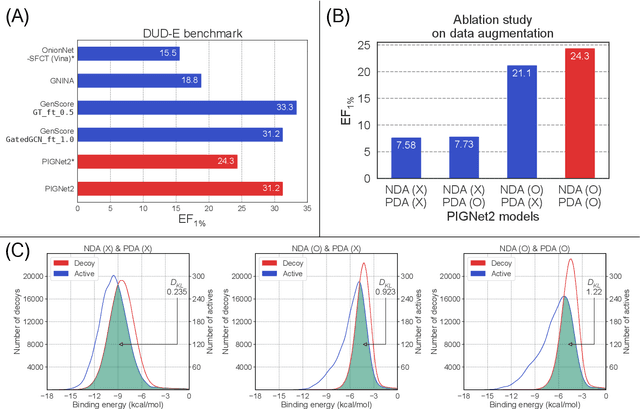
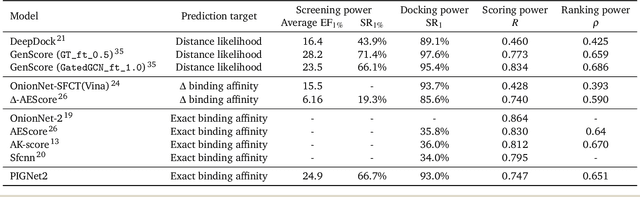
Prediction of protein-ligand interactions (PLI) plays a crucial role in drug discovery as it guides the identification and optimization of molecules that effectively bind to target proteins. Despite remarkable advances in deep learning-based PLI prediction, the development of a versatile model capable of accurately scoring binding affinity and conducting efficient virtual screening remains a challenge. The main obstacle in achieving this lies in the scarcity of experimental structure-affinity data, which limits the generalization ability of existing models. Here, we propose a viable solution to address this challenge by introducing a novel data augmentation strategy combined with a physics-informed graph neural network. The model showed significant improvements in both scoring and screening, outperforming task-specific deep learning models in various tests including derivative benchmarks, and notably achieving results comparable to the state-of-the-art performance based on distance likelihood learning. This demonstrates the potential of this approach to drug discovery.
Scaffold-based molecular design using graph generative model
May 31, 2019
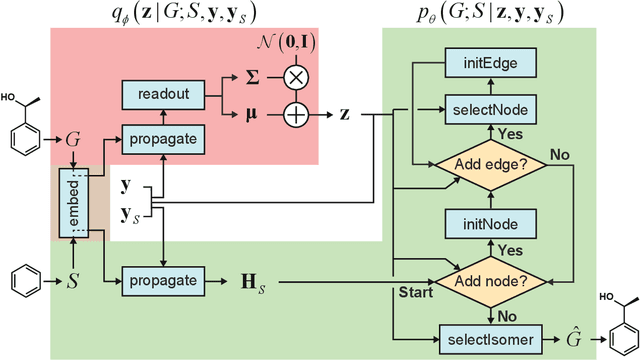
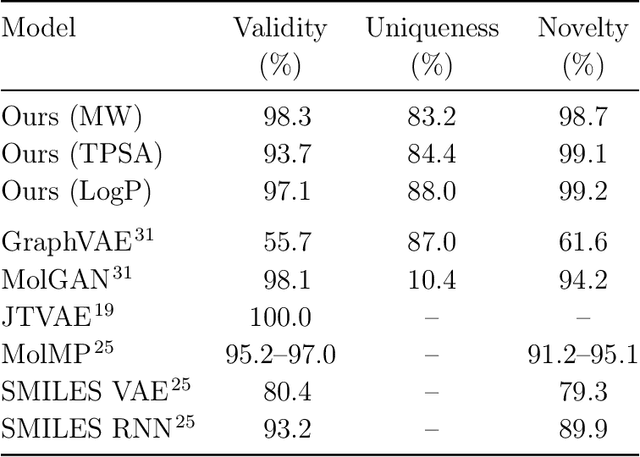
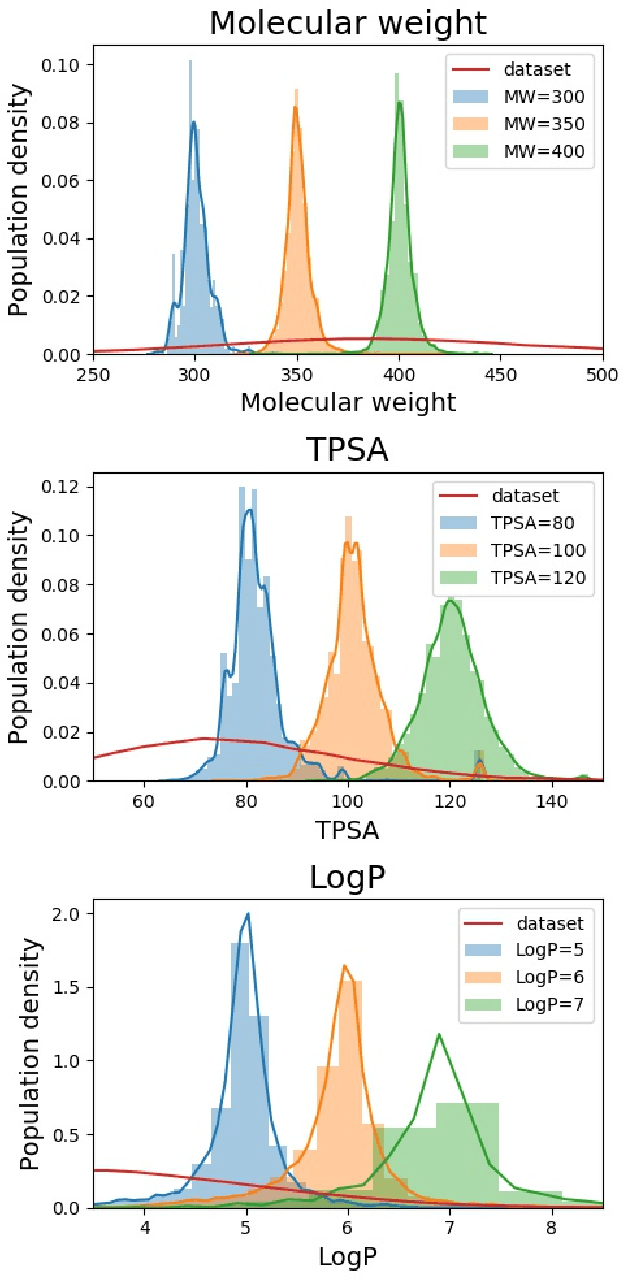
Searching new molecules in areas like drug discovery often starts from the core structures of candidate molecules to optimize the properties of interest. The way as such has called for a strategy of designing molecules retaining a particular scaffold as a substructure. On this account, our present work proposes a scaffold-based molecular generative model. The model generates molecular graphs by extending the graph of a scaffold through sequential additions of vertices and edges. In contrast to previous related models, our model guarantees the generated molecules to retain the given scaffold with certainty. Our evaluation of the model using unseen scaffolds showed the validity, uniqueness, and novelty of generated molecules as high as the case using seen scaffolds. This confirms that the model can generalize the learned chemical rules of adding atoms and bonds rather than simply memorizing the mapping from scaffolds to molecules during learning. Furthermore, despite the restraint of fixing core structures, our model could simultaneously control multiple molecular properties when generating new molecules.