 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProgressive Multi-Agent Reasoning for Biological Perturbation Prediction
Feb 07, 2026Predicting gene regulation responses to biological perturbations requires reasoning about underlying biological causalities. While large language models (LLMs) show promise for such tasks, they are often overwhelmed by the entangled nature of high-dimensional perturbation results. Moreover, recent works have primarily focused on genetic perturbations in single-cell experiments, leaving bulk-cell chemical perturbations, which is central to drug discovery, largely unexplored. Motivated by this, we present LINCSQA, a novel benchmark for predicting target gene regulation under complex chemical perturbations in bulk-cell environments. We further propose PBio-Agent, a multi-agent framework that integrates difficulty-aware task sequencing with iterative knowledge refinement. Our key insight is that genes affected by the same perturbation share causal structure, allowing confidently predicted genes to contextualize more challenging cases. The framework employs specialized agents enriched with biological knowledge graphs, while a synthesis agent integrates outputs and specialized judges ensure logical coherence. PBio-Agent outperforms existing baselines on both LINCSQA and PerturbQA, enabling even smaller models to predict and explain complex biological processes without additional training.
C3Net: interatomic potential neural network for prediction of physicochemical properties in heterogenous systems
Sep 27, 2023



Understanding the interactions of a solute with its environment is of fundamental importance in chemistry and biology. In this work, we propose a deep neural network architecture for atom type embeddings in its molecular context and interatomic potential that follows fundamental physical laws. The architecture is applied to predict physicochemical properties in heterogeneous systems including solvation in diverse solvents, 1-octanol-water partitioning, and PAMPA with a single set of network weights. We show that our architecture is generalized well to the physicochemical properties and outperforms state-of-the-art approaches based on quantum mechanics and neural networks in the task of solvation free energy prediction. The interatomic potentials at each atom in a solute obtained from the model allow quantitative analysis of the physicochemical properties at atomic resolution consistent with chemical and physical reasoning. The software is available at https://github.com/SehanLee/C3Net.
PIGNet2: A Versatile Deep Learning-based Protein-Ligand Interaction Prediction Model for Binding Affinity Scoring and Virtual Screening
Jul 17, 2023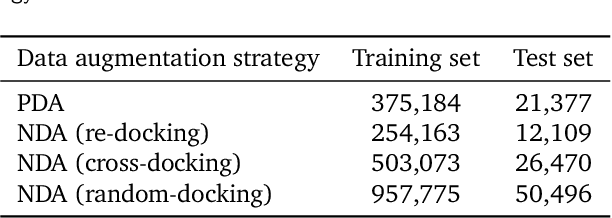
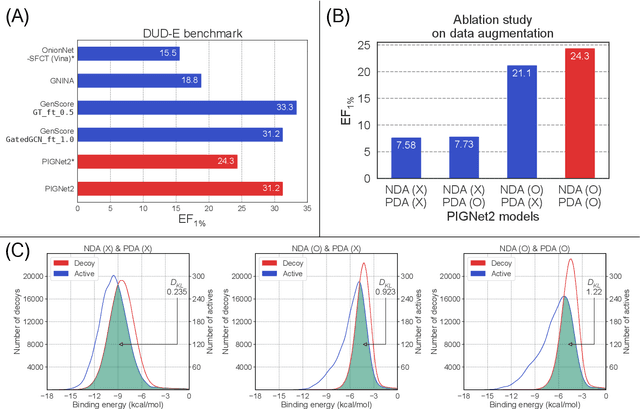
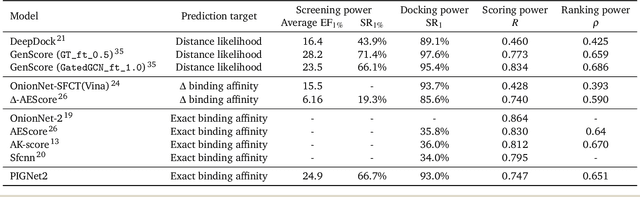
Prediction of protein-ligand interactions (PLI) plays a crucial role in drug discovery as it guides the identification and optimization of molecules that effectively bind to target proteins. Despite remarkable advances in deep learning-based PLI prediction, the development of a versatile model capable of accurately scoring binding affinity and conducting efficient virtual screening remains a challenge. The main obstacle in achieving this lies in the scarcity of experimental structure-affinity data, which limits the generalization ability of existing models. Here, we propose a viable solution to address this challenge by introducing a novel data augmentation strategy combined with a physics-informed graph neural network. The model showed significant improvements in both scoring and screening, outperforming task-specific deep learning models in various tests including derivative benchmarks, and notably achieving results comparable to the state-of-the-art performance based on distance likelihood learning. This demonstrates the potential of this approach to drug discovery.
Fragment-based molecular generative model with high generalization ability and synthetic accessibility
Nov 25, 2021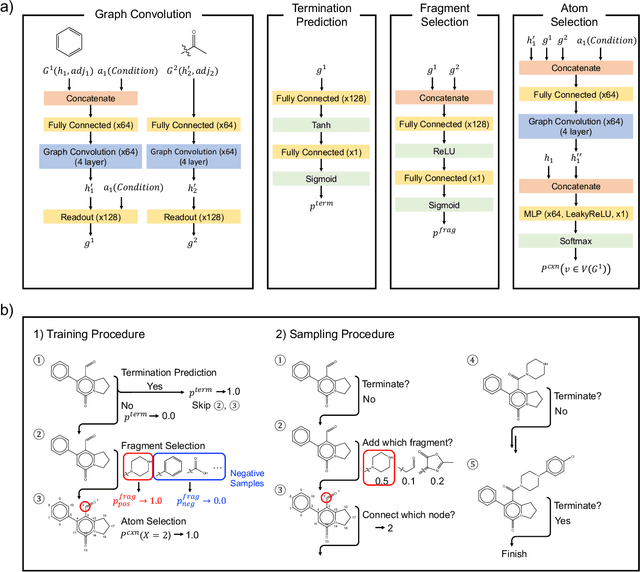
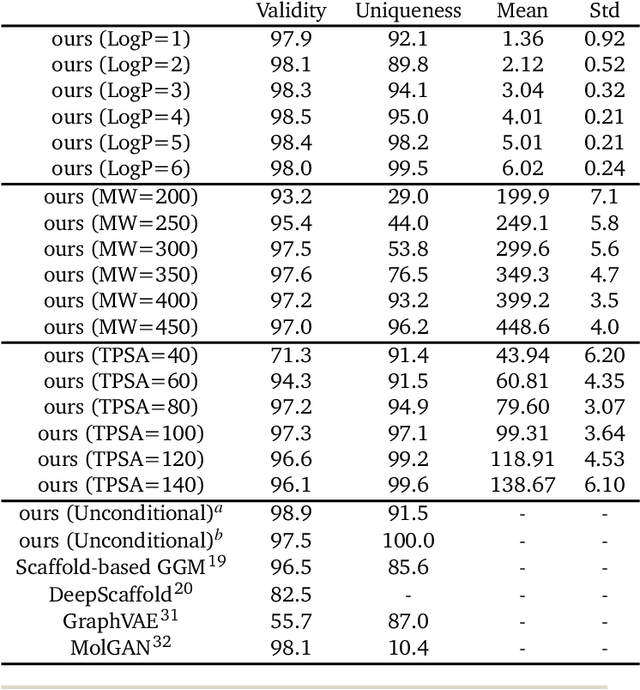
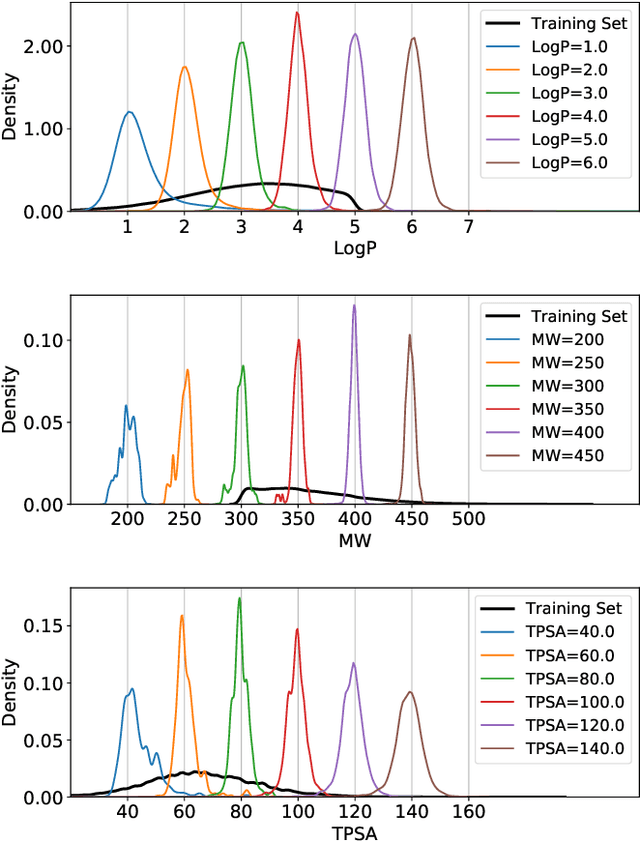
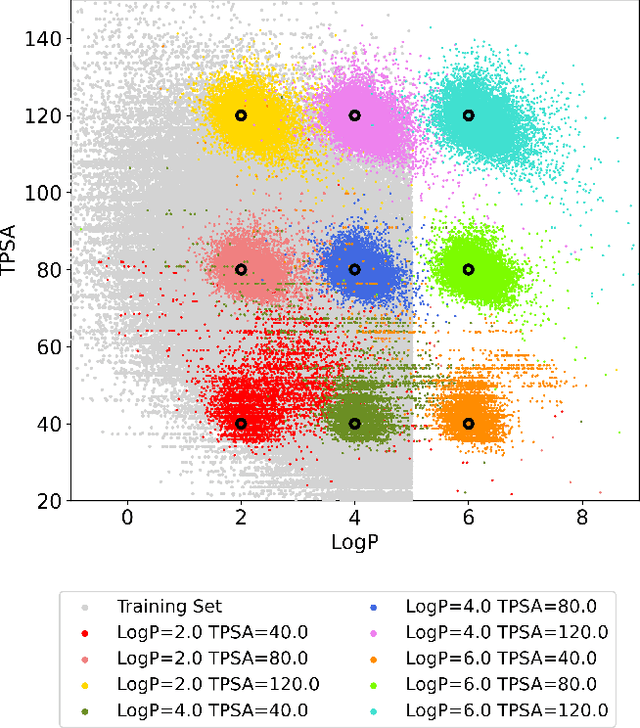
Deep generative models are attracting great attention for molecular design with desired properties. Most existing models generate molecules by sequentially adding atoms. This often renders generated molecules with less correlation with target properties and low synthetic accessibility. Molecular fragments such as functional groups are more closely related to molecular properties and synthetic accessibility than atoms. Here, we propose a fragment-based molecular generative model which designs new molecules with target properties by sequentially adding molecular fragments to any given starting molecule. A key feature of our model is a high generalization ability in terms of property control and fragment types. The former becomes possible by learning the contribution of individual fragments to the target properties in an auto-regressive manner. For the latter, we used a deep neural network that predicts the bonding probability of two molecules from the embedding vectors of the two molecules as input. The high synthetic accessibility of the generated molecules is implicitly considered while preparing the fragment library with the BRICS decomposition method. We show that the model can generate molecules with the simultaneous control of multiple target properties at a high success rate. It also works equally well with unseen fragments even in the property range where the training data is rare, verifying the high generalization ability. As a practical application, we demonstrated that the model can generate potential inhibitors with high binding affinities against the 3CL protease of SARS-COV-2 in terms of docking score.
PIGNet: A physics-informed deep learning model toward generalized drug-target interaction predictions
Aug 22, 2020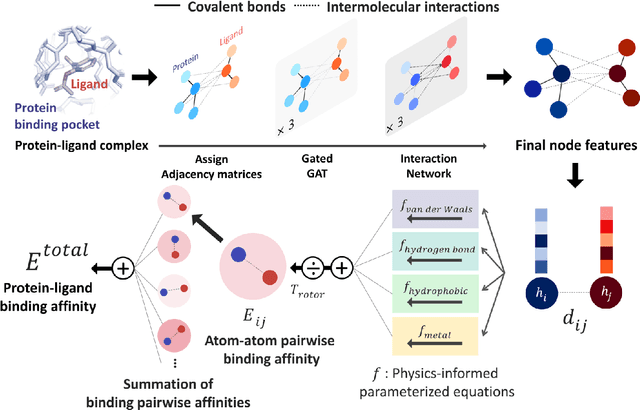
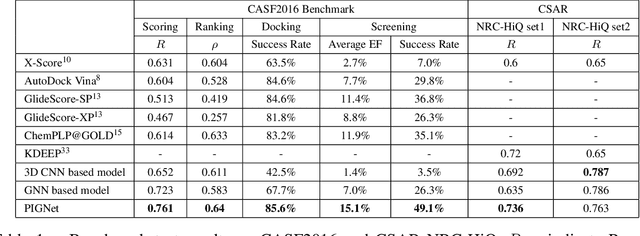
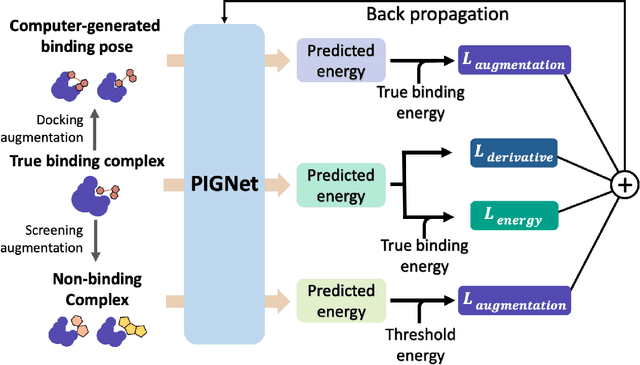
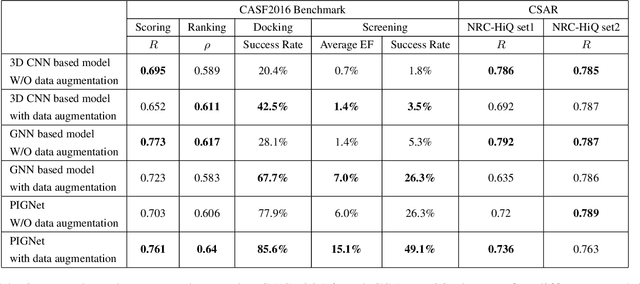
Recently, deep neural network (DNN)-based drug-target interaction (DTI) models are highlighted for their high accuracy with affordable computational costs. Yet, the models' insufficient generalization remains a challenging problem in the practice of in-silico drug discovery. We propose two key strategies to enhance generalization in the DTI model. The first one is to integrate physical models into DNN models. Our model, PIGNet, predicts the atom-atom pairwise interactions via physics-informed equations parameterized with neural networks and provides the total binding affinity of a protein-ligand complex as their sum. We further improved the model generalization by augmenting a wider range of binding poses and ligands to training data. PIGNet achieved a significant improvement in docking success rate, screening enhancement factor, and screening success rate by up to 2.01, 10.78, 14.0 times, respectively, compared to the previous DNN models. The physics-informed model also enables the interpretation of predicted binding affinities by visualizing the energy contribution of ligand substructures, providing insights for ligand optimization. Finally, we devised the uncertainty estimator of our model's prediction to qualify the outcomes and reduce the false positive rates.
Molecular Generative Model Based On Adversarially Regularized Autoencoder
Nov 13, 2019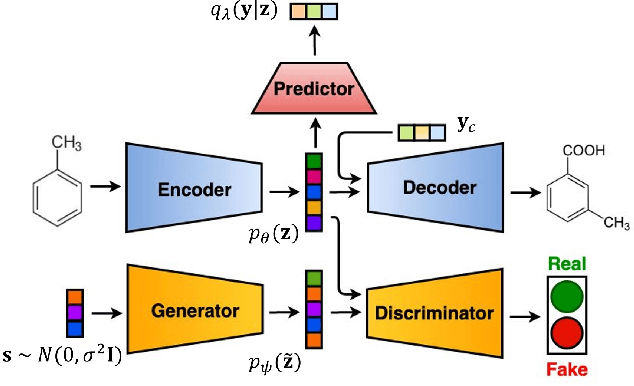
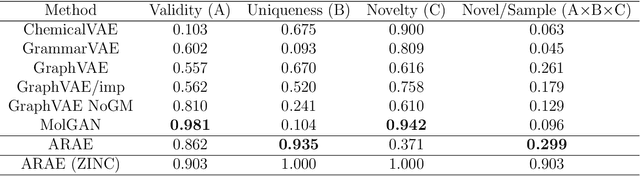
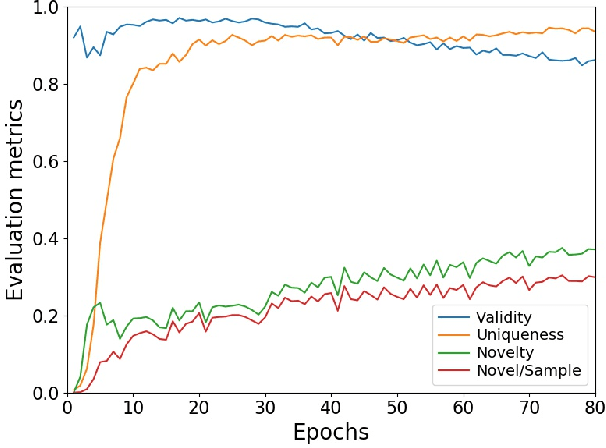
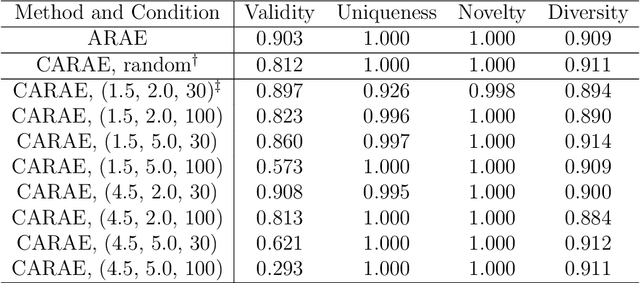
Deep generative models are attracting great attention as a new promising approach for molecular design. All models reported so far are based on either variational autoencoder (VAE) or generative adversarial network (GAN). Here we propose a new type model based on an adversarially regularized autoencoder (ARAE). It basically uses latent variables like VAE, but the distribution of the latent variables is obtained by adversarial training like in GAN. The latter is intended to avoid both inappropriate approximation of posterior distribution in VAE and difficulty in handling discrete variables in GAN. Our benchmark study showed that ARAE indeed outperformed conventional models in terms of validity, uniqueness, and novelty per generated molecule. We also demonstrated successful conditional generation of drug-like molecules with ARAE for both cases of single and multiple properties control. As a potential real-world application, we could generate EGFR inhibitors sharing the scaffolds of known active molecules while satisfying drug-like conditions simultaneously.
Scaffold-based molecular design using graph generative model
May 31, 2019
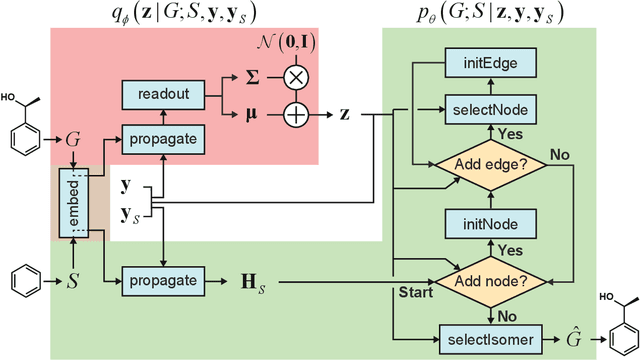
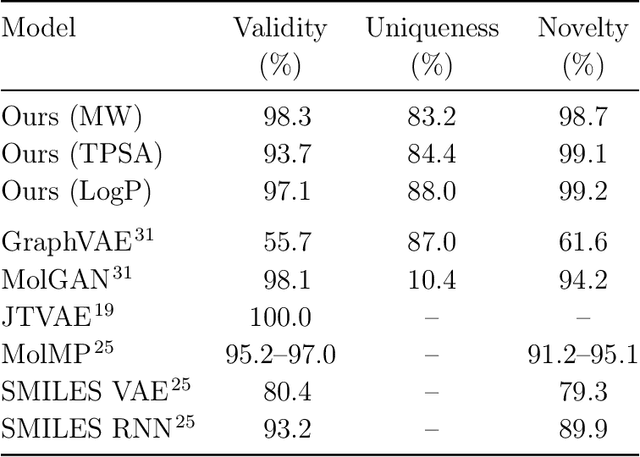
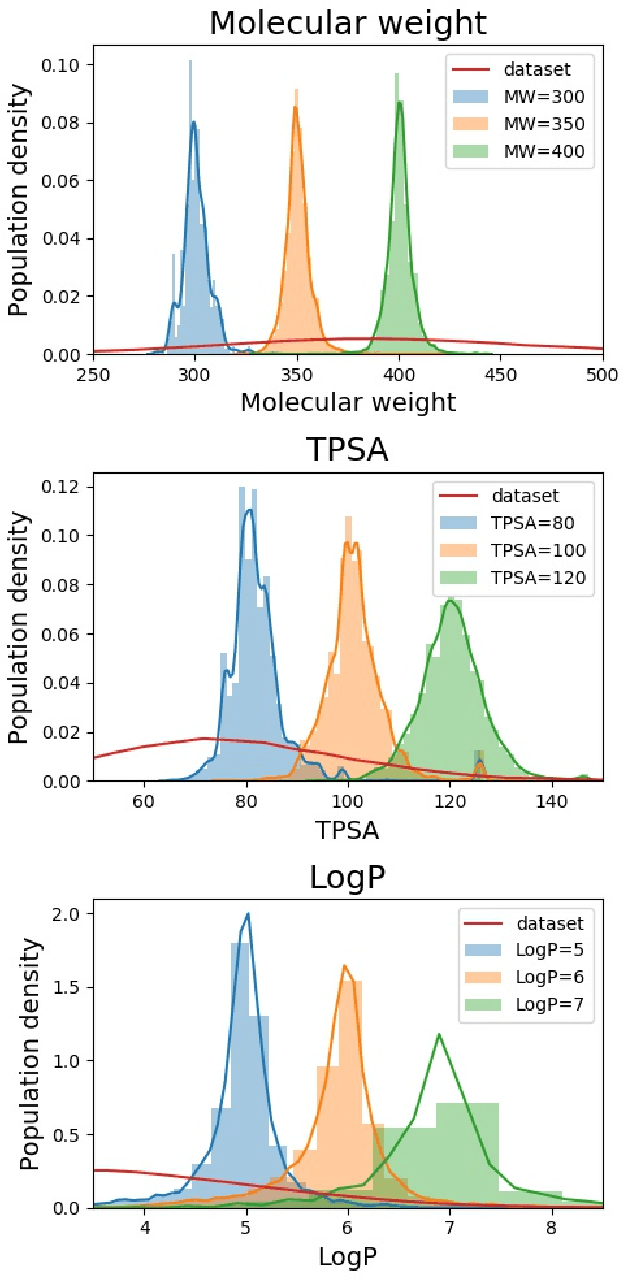
Searching new molecules in areas like drug discovery often starts from the core structures of candidate molecules to optimize the properties of interest. The way as such has called for a strategy of designing molecules retaining a particular scaffold as a substructure. On this account, our present work proposes a scaffold-based molecular generative model. The model generates molecular graphs by extending the graph of a scaffold through sequential additions of vertices and edges. In contrast to previous related models, our model guarantees the generated molecules to retain the given scaffold with certainty. Our evaluation of the model using unseen scaffolds showed the validity, uniqueness, and novelty of generated molecules as high as the case using seen scaffolds. This confirms that the model can generalize the learned chemical rules of adding atoms and bonds rather than simply memorizing the mapping from scaffolds to molecules during learning. Furthermore, despite the restraint of fixing core structures, our model could simultaneously control multiple molecular properties when generating new molecules.
Predicting drug-target interaction using 3D structure-embedded graph representations from graph neural networks
Apr 17, 2019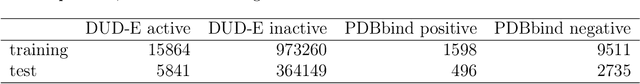
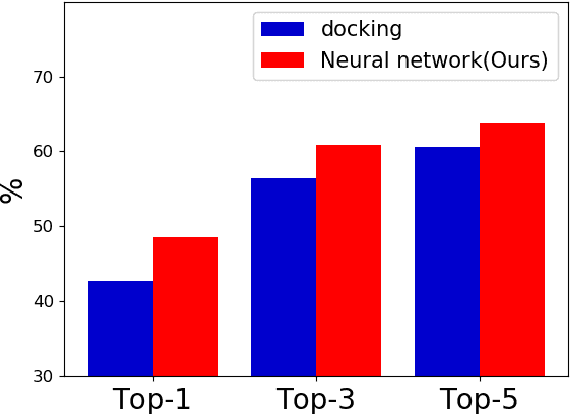

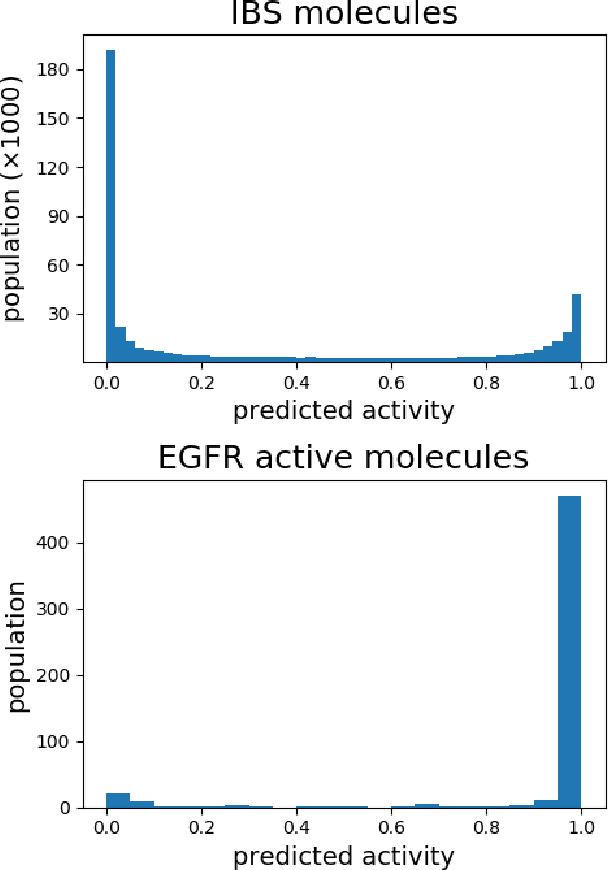
Accurate prediction of drug-target interaction (DTI) is essential for in silico drug design. For the purpose, we propose a novel approach for predicting DTI using a GNN that directly incorporates the 3D structure of a protein-ligand complex. We also apply a distance-aware graph attention algorithm with gate augmentation to increase the performance of our model. As a result, our model shows better performance than docking and other deep learning methods for both virtual screening and pose prediction. In addition, our model can reproduce the natural population distribution of active molecules and inactive molecules.
Deeply learning molecular structure-property relationships using attention- and gate-augmented graph convolutional network
Oct 08, 2018


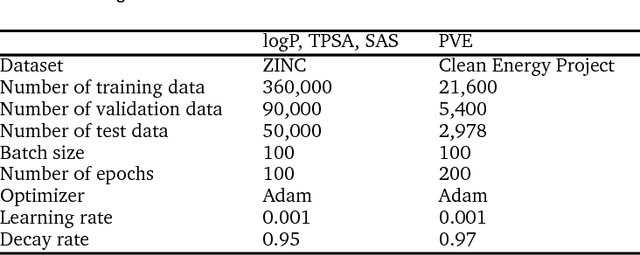
Molecular structure-property relationships are key to molecular engineering for materials and drug discovery. The rise of deep learning offers a new viable solution to elucidate the structure-property relationships directly from chemical data. Here we show that the performance of graph convolutional networks (GCNs) for the prediction of molecular properties can be improved by incorporating attention and gate mechanisms. The attention mechanism enables a GCN to identify atoms in different environments. The gated skip-connection further improves the GCN by updating feature maps at an appropriate rate. We demonstrate that the resulting attention- and gate-augmented GCN could extract better structural features related to a target molecular property such as solubility, polarity, synthetic accessibility and photovoltaic efficiency compared to the vanilla GCN. More interestingly, it identified two distinct parts of molecules as essential structural features for high photovoltaic efficiency, and each of them coincided with the areas of donor and acceptor orbitals for charge-transfer excitations, respectively. As a result, the new model could accurately predict molecular properties and place molecules with similar properties close to each other in a well-trained latent space, which is critical for successful molecular engineering.
Molecular generative model based on conditional variational autoencoder for de novo molecular design
Jun 15, 2018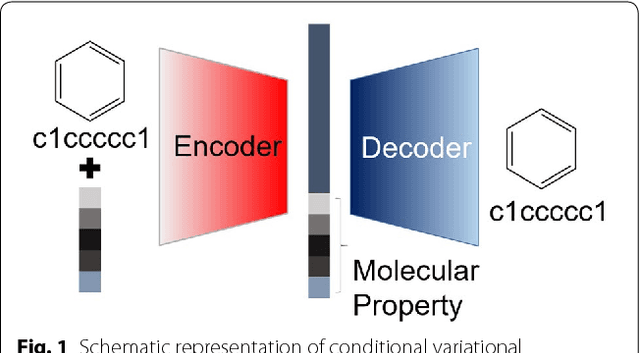

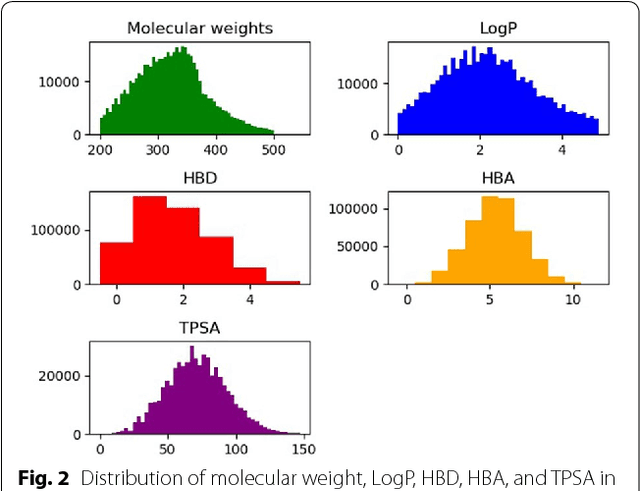

We propose a molecular generative model based on the conditional variational autoencoder for de novo molecular design. It is specialized to control multiple molecular properties simultaneously by imposing them on a latent space. As a proof of concept, we demonstrate that it can be used to generate drug-like molecules with five target properties. We were also able to adjust a single property without changing the others and to manipulate it beyond the range of the dataset.