 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecular Generative Model Based On Adversarially Regularized Autoencoder
Nov 13, 2019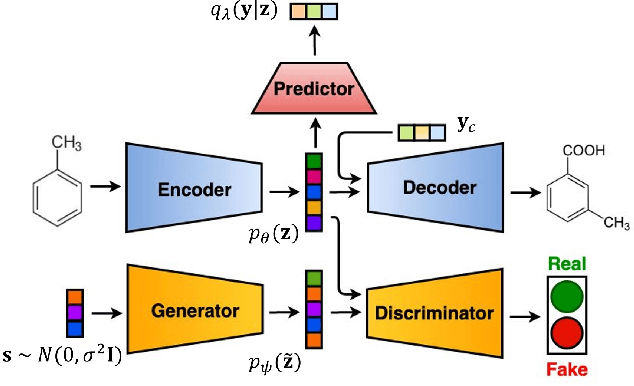
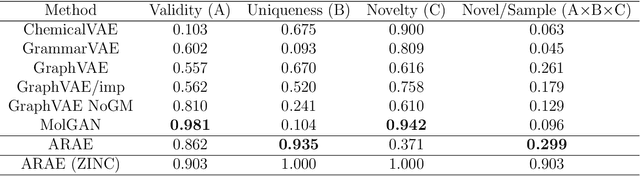
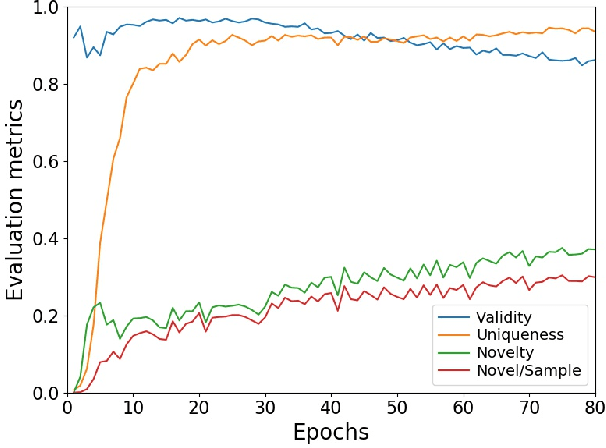
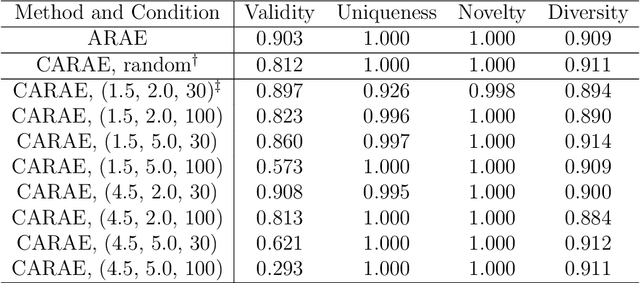
Deep generative models are attracting great attention as a new promising approach for molecular design. All models reported so far are based on either variational autoencoder (VAE) or generative adversarial network (GAN). Here we propose a new type model based on an adversarially regularized autoencoder (ARAE). It basically uses latent variables like VAE, but the distribution of the latent variables is obtained by adversarial training like in GAN. The latter is intended to avoid both inappropriate approximation of posterior distribution in VAE and difficulty in handling discrete variables in GAN. Our benchmark study showed that ARAE indeed outperformed conventional models in terms of validity, uniqueness, and novelty per generated molecule. We also demonstrated successful conditional generation of drug-like molecules with ARAE for both cases of single and multiple properties control. As a potential real-world application, we could generate EGFR inhibitors sharing the scaffolds of known active molecules while satisfying drug-like conditions simultaneously.
Deeply learning molecular structure-property relationships using attention- and gate-augmented graph convolutional network
Oct 08, 2018


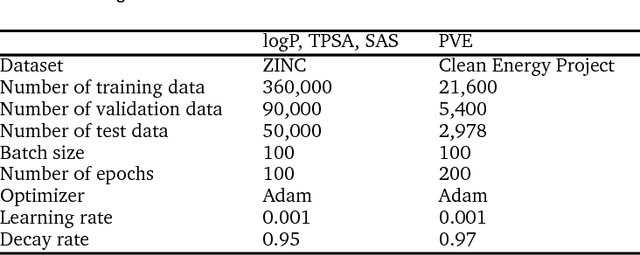
Molecular structure-property relationships are key to molecular engineering for materials and drug discovery. The rise of deep learning offers a new viable solution to elucidate the structure-property relationships directly from chemical data. Here we show that the performance of graph convolutional networks (GCNs) for the prediction of molecular properties can be improved by incorporating attention and gate mechanisms. The attention mechanism enables a GCN to identify atoms in different environments. The gated skip-connection further improves the GCN by updating feature maps at an appropriate rate. We demonstrate that the resulting attention- and gate-augmented GCN could extract better structural features related to a target molecular property such as solubility, polarity, synthetic accessibility and photovoltaic efficiency compared to the vanilla GCN. More interestingly, it identified two distinct parts of molecules as essential structural features for high photovoltaic efficiency, and each of them coincided with the areas of donor and acceptor orbitals for charge-transfer excitations, respectively. As a result, the new model could accurately predict molecular properties and place molecules with similar properties close to each other in a well-trained latent space, which is critical for successful molecular engineering.