 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCompositional Flows for 3D Molecule and Synthesis Pathway Co-design
Apr 10, 2025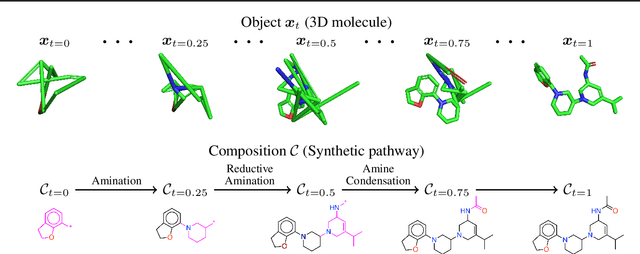
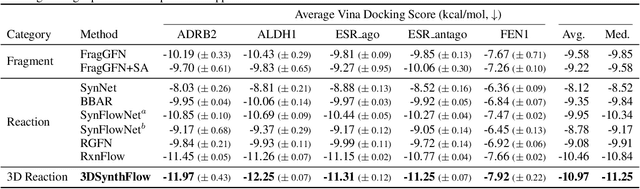
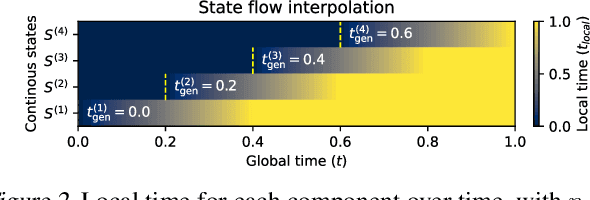
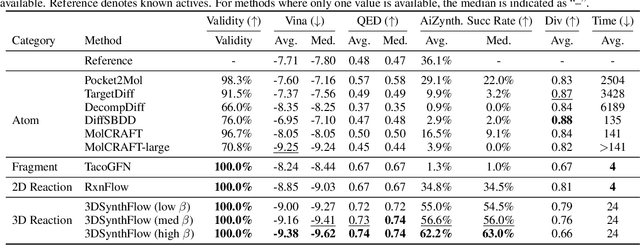
Many generative applications, such as synthesis-based 3D molecular design, involve constructing compositional objects with continuous features. Here, we introduce Compositional Generative Flows (CGFlow), a novel framework that extends flow matching to generate objects in compositional steps while modeling continuous states. Our key insight is that modeling compositional state transitions can be formulated as a straightforward extension of the flow matching interpolation process. We further build upon the theoretical foundations of generative flow networks (GFlowNets), enabling reward-guided sampling of compositional structures. We apply CGFlow to synthesizable drug design by jointly designing the molecule's synthetic pathway with its 3D binding pose. Our approach achieves state-of-the-art binding affinity on all 15 targets from the LIT-PCBA benchmark, and 5.8$\times$ improvement in sampling efficiency compared to 2D synthesis-based baseline. To our best knowledge, our method is also the first to achieve state of-art-performance in both Vina Dock (-9.38) and AiZynth success rate (62.2\%) on the CrossDocked benchmark.
Generative Flows on Synthetic Pathway for Drug Design
Oct 06, 2024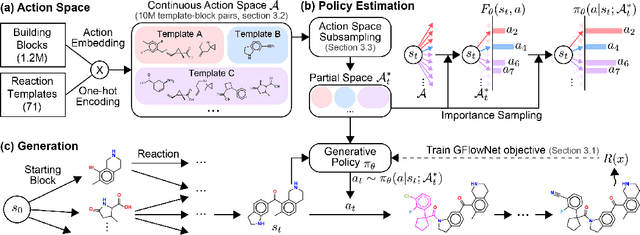
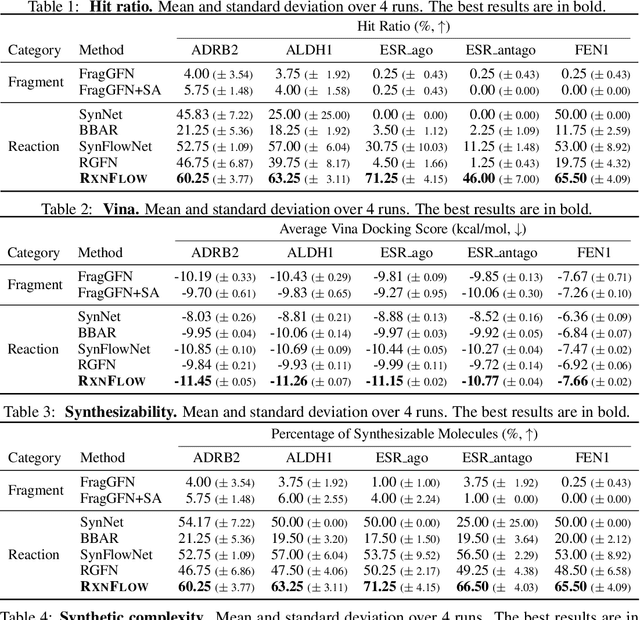
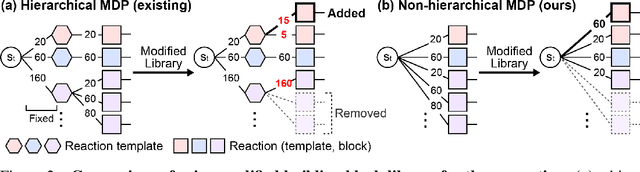
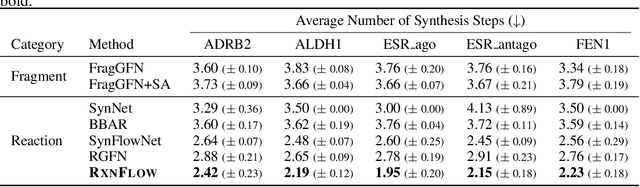
Generative models in drug discovery have recently gained attention as efficient alternatives to brute-force virtual screening. However, most existing models do not account for synthesizability, limiting their practical use in real-world scenarios. In this paper, we propose RxnFlow, which sequentially assembles molecules using predefined molecular building blocks and chemical reaction templates to constrain the synthetic chemical pathway. We then train on this sequential generating process with the objective of generative flow networks (GFlowNets) to generate both highly rewarded and diverse molecules. To mitigate the large action space of synthetic pathways in GFlowNets, we implement a novel action space subsampling method. This enables RxnFlow to learn generative flows over extensive action spaces comprising combinations of 1.2 million building blocks and 71 reaction templates without significant computational overhead. Additionally, RxnFlow can employ modified or expanded action spaces for generation without retraining, allowing for the introduction of additional objectives or the incorporation of newly discovered building blocks. We experimentally demonstrate that RxnFlow outperforms existing reaction-based and fragment-based models in pocket-specific optimization across various target pockets. Furthermore, RxnFlow achieves state-of-the-art performance on CrossDocked2020 for pocket-conditional generation, with an average Vina score of -8.85kcal/mol and 34.8% synthesizability.
PharmacoNet: Accelerating Large-Scale Virtual Screening by Deep Pharmacophore Modeling
Oct 04, 2023



As the size of accessible compound libraries expands to over 10 billion, the need for more efficient structure-based virtual screening methods is emerging. Different pre-screening methods have been developed to rapidly screen the library, but the structure-based methods applicable to general proteins are still lacking: the challenge is to predict the binding pose between proteins and ligands and perform scoring in an extremely short time. We introduce PharmacoNet, a deep learning framework that identifies the optimal 3D pharmacophore arrangement which a ligand should have for stable binding from the binding site. By coarse-grained graph matching between ligands and the generated pharmacophore arrangement, we solve the expensive binding pose sampling and scoring procedures of existing methods in a single step. PharmacoNet is significantly faster than state-of-the-art structure-based approaches, yet reasonably accurate with a simple scoring function. Furthermore, we show the promising result that PharmacoNet effectively retains hit candidates even under the high pre-screening filtration rates. Overall, our study uncovers the hitherto untapped potential of a pharmacophore modeling approach in deep learning-based drug discovery.
Fragment-based molecular generative model with high generalization ability and synthetic accessibility
Nov 25, 2021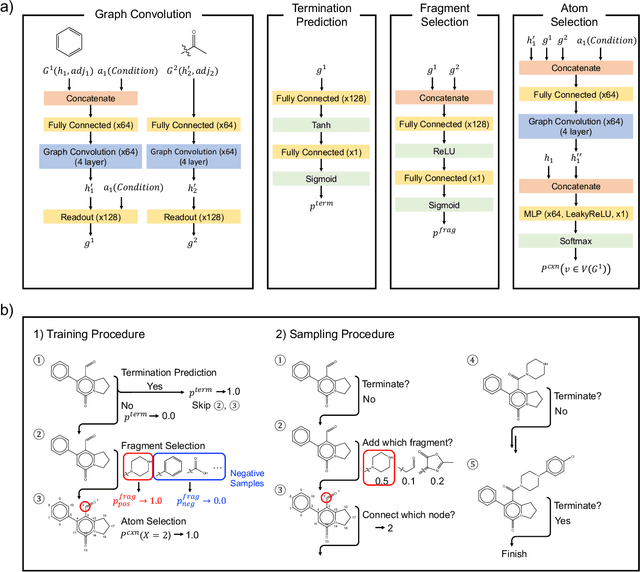
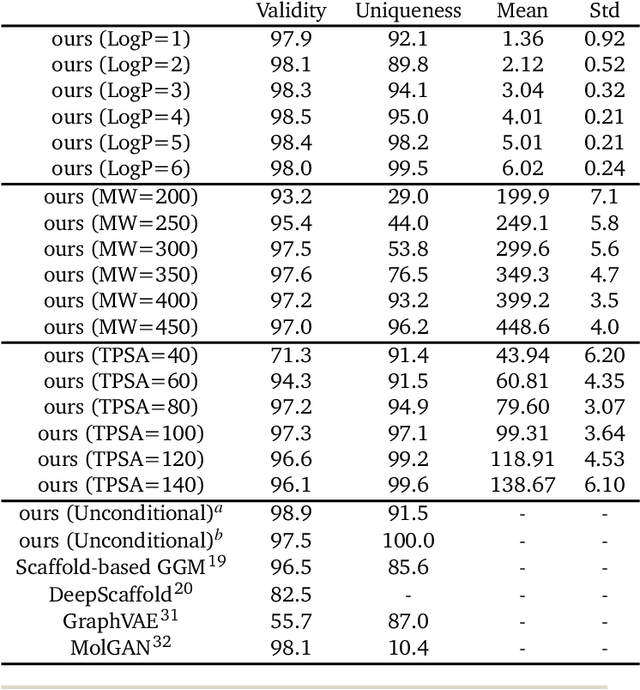
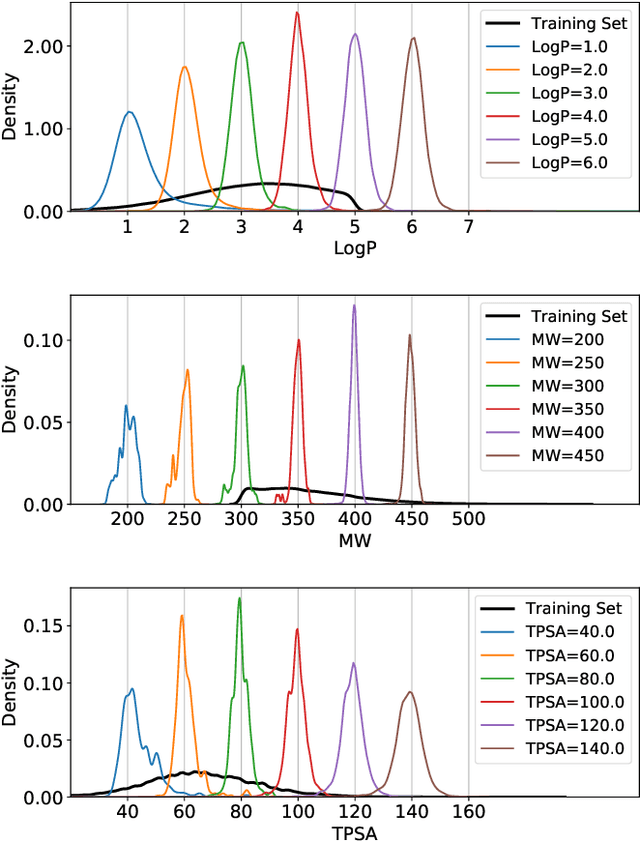
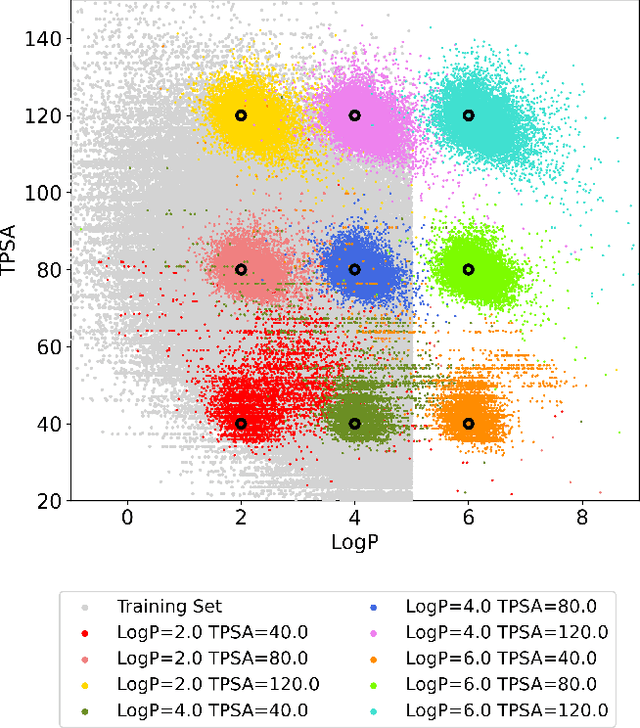
Deep generative models are attracting great attention for molecular design with desired properties. Most existing models generate molecules by sequentially adding atoms. This often renders generated molecules with less correlation with target properties and low synthetic accessibility. Molecular fragments such as functional groups are more closely related to molecular properties and synthetic accessibility than atoms. Here, we propose a fragment-based molecular generative model which designs new molecules with target properties by sequentially adding molecular fragments to any given starting molecule. A key feature of our model is a high generalization ability in terms of property control and fragment types. The former becomes possible by learning the contribution of individual fragments to the target properties in an auto-regressive manner. For the latter, we used a deep neural network that predicts the bonding probability of two molecules from the embedding vectors of the two molecules as input. The high synthetic accessibility of the generated molecules is implicitly considered while preparing the fragment library with the BRICS decomposition method. We show that the model can generate molecules with the simultaneous control of multiple target properties at a high success rate. It also works equally well with unseen fragments even in the property range where the training data is rare, verifying the high generalization ability. As a practical application, we demonstrated that the model can generate potential inhibitors with high binding affinities against the 3CL protease of SARS-COV-2 in terms of docking score.