 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProtein Language Model Embeddings Improve Generalization of Implicit Transfer Operators
Feb 11, 2026Molecular dynamics (MD) is a central computational tool in physics, chemistry, and biology, enabling quantitative prediction of experimental observables as expectations over high-dimensional molecular distributions such as Boltzmann distributions and transition densities. However, conventional MD is fundamentally limited by the high computational cost required to generate independent samples. Generative molecular dynamics (GenMD) has recently emerged as an alternative, learning surrogates of molecular distributions either from data or through interaction with energy models. While these methods enable efficient sampling, their transferability across molecular systems is often limited. In this work, we show that incorporating auxiliary sources of information can improve the data efficiency and generalization of transferable implicit transfer operators (TITO) for molecular dynamics. We find that coarse-grained TITO models are substantially more data-efficient than Boltzmann Emulators, and that incorporating protein language model (pLM) embeddings further improves out-of-distribution generalization. Our approach, PLaTITO, achieves state-of-the-art performance on equilibrium sampling benchmarks for out-of-distribution protein systems, including fast-folding proteins. We further study the impact of additional conditioning signals -- such as structural embeddings, temperature, and large-language-model-derived embeddings -- on model performance.
HollowFlow: Efficient Sample Likelihood Evaluation using Hollow Message Passing
Oct 24, 2025



Flow and diffusion-based models have emerged as powerful tools for scientific applications, particularly for sampling non-normalized probability distributions, as exemplified by Boltzmann Generators (BGs). A critical challenge in deploying these models is their reliance on sample likelihood computations, which scale prohibitively with system size $n$, often rendering them infeasible for large-scale problems. To address this, we introduce $\textit{HollowFlow}$, a flow-based generative model leveraging a novel non-backtracking graph neural network (NoBGNN). By enforcing a block-diagonal Jacobian structure, HollowFlow likelihoods are evaluated with a constant number of backward passes in $n$, yielding speed-ups of up to $\mathcal{O}(n^2)$: a significant step towards scaling BGs to larger systems. Crucially, our framework generalizes: $\textbf{any equivariant GNN or attention-based architecture}$ can be adapted into a NoBGNN. We validate HollowFlow by training BGs on two different systems of increasing size. For both systems, the sampling and likelihood evaluation time decreases dramatically, following our theoretical scaling laws. For the larger system we obtain a $10^2\times$ speed-up, clearly illustrating the potential of HollowFlow-based approaches for high-dimensional scientific problems previously hindered by computational bottlenecks.
Generative flow-based warm start of the variational quantum eigensolver
Jul 02, 2025Hybrid quantum-classical algorithms like the variational quantum eigensolver (VQE) show promise for quantum simulations on near-term quantum devices, but are often limited by complex objective functions and expensive optimization procedures. Here, we propose Flow-VQE, a generative framework leveraging conditional normalizing flows with parameterized quantum circuits to efficiently generate high-quality variational parameters. By embedding a generative model into the VQE optimization loop through preference-based training, Flow-VQE enables quantum gradient-free optimization and offers a systematic approach for parameter transfer, accelerating convergence across related problems through warm-started optimization. We compare Flow-VQE to a number of standard benchmarks through numerical simulations on molecular systems, including hydrogen chains, water, ammonia, and benzene. We find that Flow-VQE outperforms baseline optimization algorithms, achieving computational accuracy with fewer circuit evaluations (improvements range from modest to more than two orders of magnitude) and, when used to warm-start the optimization of new systems, accelerates subsequent fine-tuning by up to 50-fold compared with Hartree--Fock initialization. Therefore, we believe Flow-VQE can become a pragmatic and versatile paradigm for leveraging generative modeling to reduce the costs of variational quantum algorithms.
Minimum-Excess-Work Guidance
May 19, 2025We propose a regularization framework inspired by thermodynamic work for guiding pre-trained probability flow generative models (e.g., continuous normalizing flows or diffusion models) by minimizing excess work, a concept rooted in statistical mechanics and with strong conceptual connections to optimal transport. Our approach enables efficient guidance in sparse-data regimes common to scientific applications, where only limited target samples or partial density constraints are available. We introduce two strategies: Path Guidance for sampling rare transition states by concentrating probability mass on user-defined subsets, and Observable Guidance for aligning generated distributions with experimental observables while preserving entropy. We demonstrate the framework's versatility on a coarse-grained protein model, guiding it to sample transition configurations between folded/unfolded states and correct systematic biases using experimental data. The method bridges thermodynamic principles with modern generative architectures, offering a principled, efficient, and physics-inspired alternative to standard fine-tuning in data-scarce domains. Empirical results highlight improved sample efficiency and bias reduction, underscoring its applicability to molecular simulations and beyond.
FLOWR: Flow Matching for Structure-Aware De Novo, Interaction- and Fragment-Based Ligand Generation
Apr 14, 2025
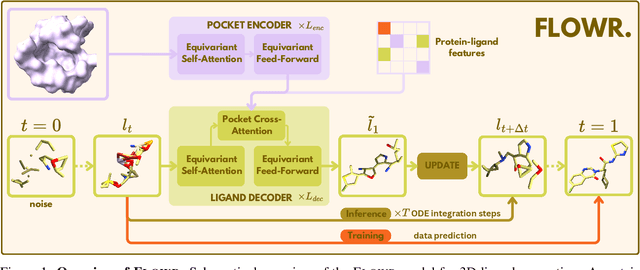
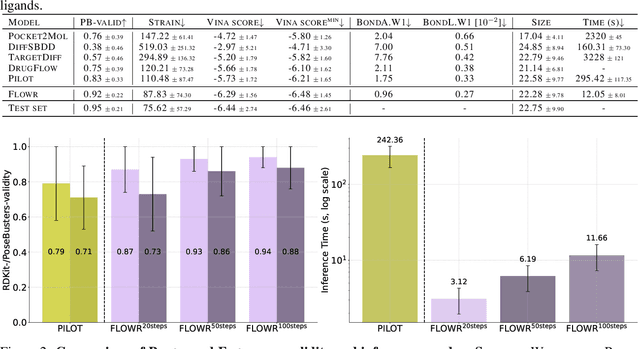
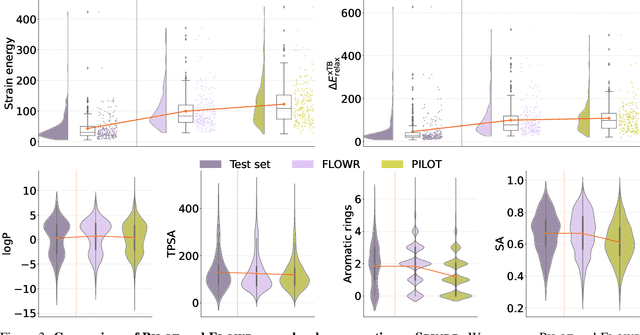
We introduce FLOWR, a novel structure-based framework for the generation and optimization of three-dimensional ligands. FLOWR integrates continuous and categorical flow matching with equivariant optimal transport, enhanced by an efficient protein pocket conditioning. Alongside FLOWR, we present SPINDR, a thoroughly curated dataset comprising ligand-pocket co-crystal complexes specifically designed to address existing data quality issues. Empirical evaluations demonstrate that FLOWR surpasses current state-of-the-art diffusion- and flow-based methods in terms of PoseBusters-validity, pose accuracy, and interaction recovery, while offering a significant inference speedup, achieving up to 70-fold faster performance. In addition, we introduce FLOWR.multi, a highly accurate multi-purpose model allowing for the targeted sampling of novel ligands that adhere to predefined interaction profiles and chemical substructures for fragment-based design without the need of re-training or any re-sampling strategies
Compositional Flows for 3D Molecule and Synthesis Pathway Co-design
Apr 10, 2025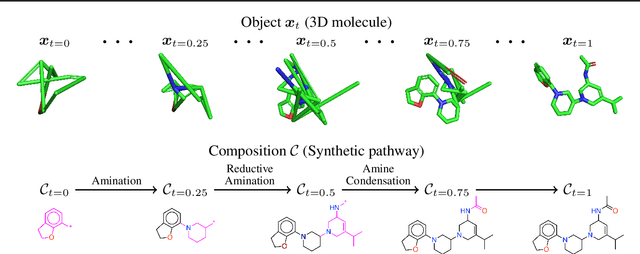
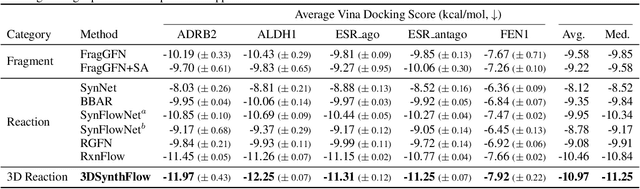
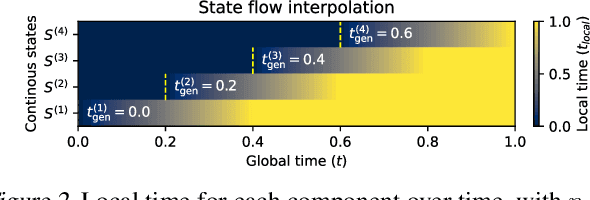
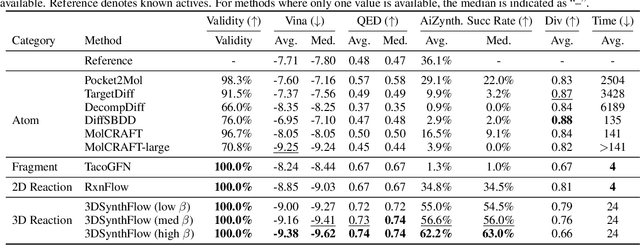
Many generative applications, such as synthesis-based 3D molecular design, involve constructing compositional objects with continuous features. Here, we introduce Compositional Generative Flows (CGFlow), a novel framework that extends flow matching to generate objects in compositional steps while modeling continuous states. Our key insight is that modeling compositional state transitions can be formulated as a straightforward extension of the flow matching interpolation process. We further build upon the theoretical foundations of generative flow networks (GFlowNets), enabling reward-guided sampling of compositional structures. We apply CGFlow to synthesizable drug design by jointly designing the molecule's synthetic pathway with its 3D binding pose. Our approach achieves state-of-the-art binding affinity on all 15 targets from the LIT-PCBA benchmark, and 5.8$\times$ improvement in sampling efficiency compared to 2D synthesis-based baseline. To our best knowledge, our method is also the first to achieve state of-art-performance in both Vina Dock (-9.38) and AiZynth success rate (62.2\%) on the CrossDocked benchmark.
Efficient 3D Molecular Generation with Flow Matching and Scale Optimal Transport
Jun 11, 2024



Generative models for 3D drug design have gained prominence recently for their potential to design ligands directly within protein pockets. Current approaches, however, often suffer from very slow sampling times or generate molecules with poor chemical validity. Addressing these limitations, we propose Semla, a scalable E(3)-equivariant message passing architecture. We further introduce a molecular generation model, MolFlow, which is trained using flow matching along with scale optimal transport, a novel extension of equivariant optimal transport. Our model produces state-of-the-art results on benchmark datasets with just 100 sampling steps. Crucially, MolFlow samples high quality molecules with as few as 20 steps, corresponding to a two order-of-magnitude speed-up compared to state-of-the-art, without sacrificing performance. Furthermore, we highlight limitations of current evaluation methods for 3D generation and propose new benchmark metrics for unconditional molecular generators. Finally, using these new metrics, we compare our model's ability to generate high quality samples against current approaches and further demonstrate MolFlow's strong performance.
Navigating protein landscapes with a machine-learned transferable coarse-grained model
Oct 27, 2023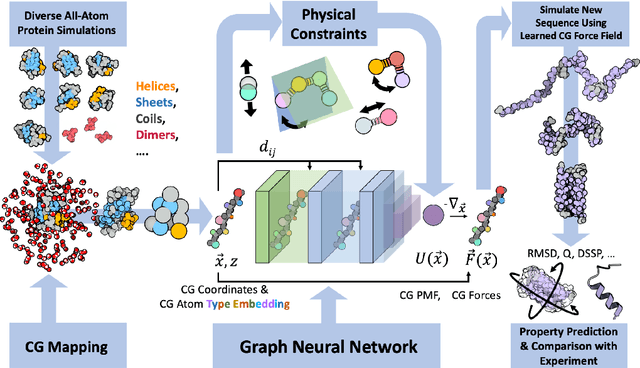
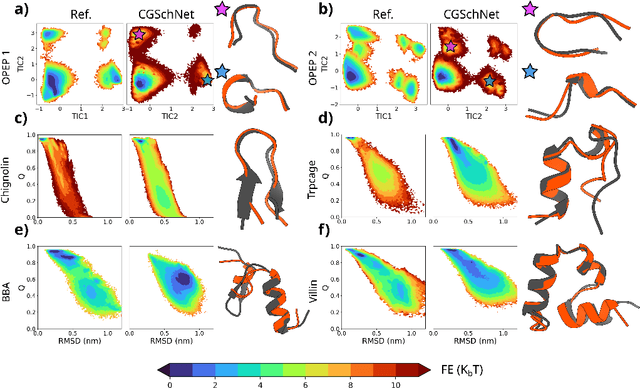

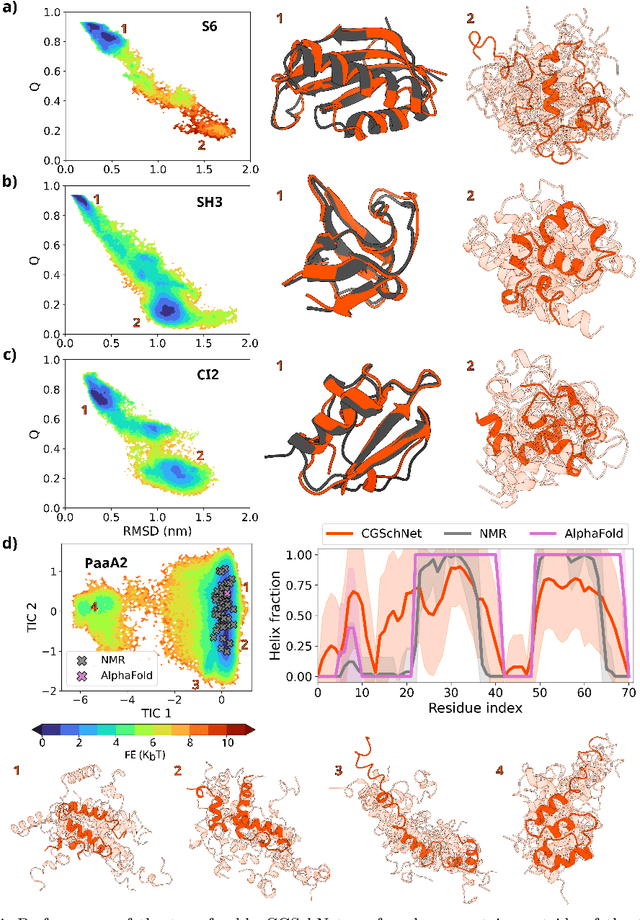
The most popular and universally predictive protein simulation models employ all-atom molecular dynamics (MD), but they come at extreme computational cost. The development of a universal, computationally efficient coarse-grained (CG) model with similar prediction performance has been a long-standing challenge. By combining recent deep learning methods with a large and diverse training set of all-atom protein simulations, we here develop a bottom-up CG force field with chemical transferability, which can be used for extrapolative molecular dynamics on new sequences not used during model parametrization. We demonstrate that the model successfully predicts folded structures, intermediates, metastable folded and unfolded basins, and the fluctuations of intrinsically disordered proteins while it is several orders of magnitude faster than an all-atom model. This showcases the feasibility of a universal and computationally efficient machine-learned CG model for proteins.
Implicit Transfer Operator Learning: Multiple Time-Resolution Surrogates for Molecular Dynamics
May 29, 2023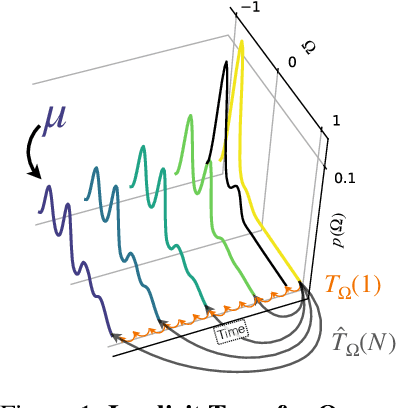

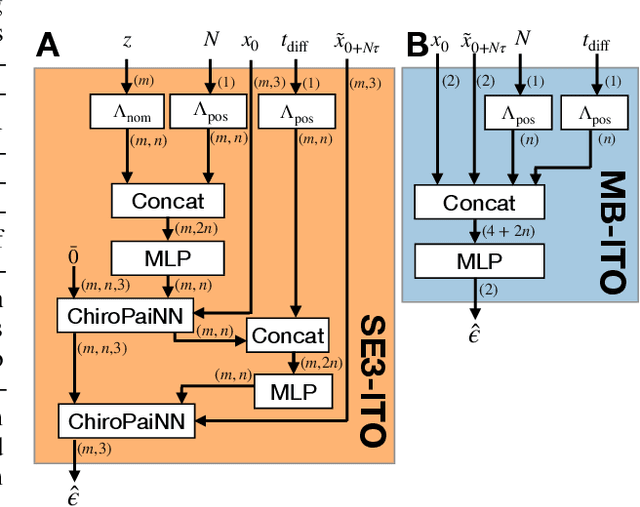

Computing properties of molecular systems rely on estimating expectations of the (unnormalized) Boltzmann distribution. Molecular dynamics (MD) is a broadly adopted technique to approximate such quantities. However, stable simulations rely on very small integration time-steps ($10^{-15}\,\mathrm{s}$), whereas convergence of some moments, e.g. binding free energy or rates, might rely on sampling processes on time-scales as long as $10^{-1}\, \mathrm{s}$, and these simulations must be repeated for every molecular system independently. Here, we present Implict Transfer Operator (ITO) Learning, a framework to learn surrogates of the simulation process with multiple time-resolutions. We implement ITO with denoising diffusion probabilistic models with a new SE(3) equivariant architecture and show the resulting models can generate self-consistent stochastic dynamics across multiple time-scales, even when the system is only partially observed. Finally, we present a coarse-grained CG-SE3-ITO model which can quantitatively model all-atom molecular dynamics using only coarse molecular representations. As such, ITO provides an important step towards multiple time- and space-resolution acceleration of MD.
Coarse Graining Molecular Dynamics with Graph Neural Networks
Aug 21, 2020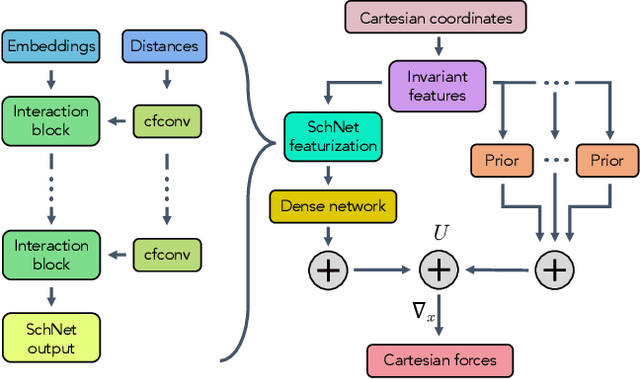
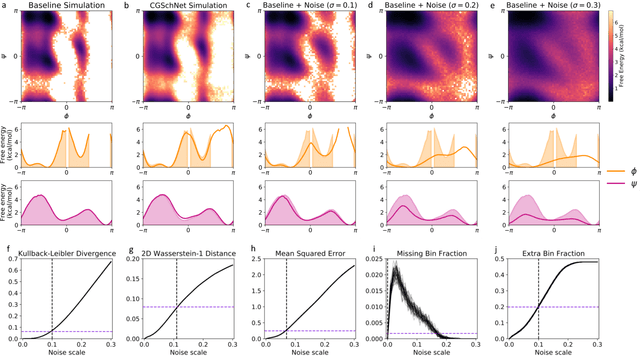
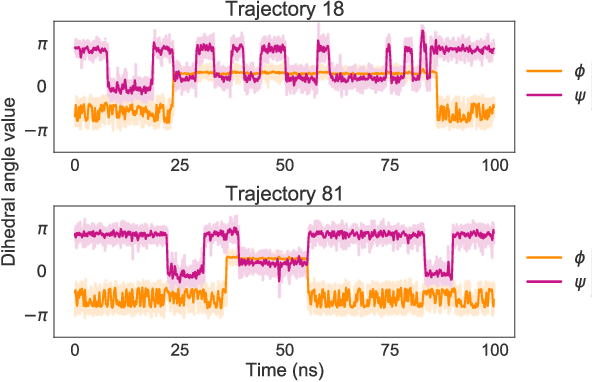
Coarse graining enables the investigation of molecular dynamics for larger systems and at longer timescales than is possible at atomic resolution. However, a coarse graining model must be formulated such that the conclusions we draw from it are consistent with the conclusions we would draw from a model at a finer level of detail. It has been proven that a force matching scheme defines a thermodynamically consistent coarse-grained model for an atomistic system in the variational limit. Wang et al. [ACS Cent. Sci. 5, 755 (2019)] demonstrated that the existence of such a variational limit enables the use of a supervised machine learning framework to generate a coarse-grained force field, which can then be used for simulation in the coarse-grained space. Their framework, however, requires the manual input of molecular features upon which to machine learn the force field. In the present contribution, we build upon the advance of Wang et al.and introduce a hybrid architecture for the machine learning of coarse-grained force fields that learns their own features via a subnetwork that leverages continuous filter convolutions on a graph neural network architecture. We demonstrate that this framework succeeds at reproducing the thermodynamics for small biomolecular systems. Since the learned molecular representations are inherently transferable, the architecture presented here sets the stage for the development of machine-learned, coarse-grained force fields that are transferable across molecular systems.