 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScaling Atomistic Protein Binder Design with Generative Pretraining and Test-Time Compute
Mar 30, 2026Protein interaction modeling is central to protein design, which has been transformed by machine learning with applications in drug discovery and beyond. In this landscape, structure-based de novo binder design is cast as either conditional generative modeling or sequence optimization via structure predictors ("hallucination"). We argue that this is a false dichotomy and propose Proteina-Complexa, a novel fully atomistic binder generation method unifying both paradigms. We extend recent flow-based latent protein generation architectures and leverage the domain-domain interactions of monomeric computationally predicted protein structures to construct Teddymer, a new large-scale dataset of synthetic binder-target pairs for pretraining. Combined with high-quality experimental multimers, this enables training a strong base model. We then perform inference-time optimization with this generative prior, unifying the strengths of previously distinct generative and hallucination methods. Proteina-Complexa sets a new state of the art in computational binder design benchmarks: it delivers markedly higher in-silico success rates than existing generative approaches, and our novel test-time optimization strategies greatly outperform previous hallucination methods under normalized compute budgets. We also demonstrate interface hydrogen bond optimization, fold class-guided binder generation, and extensions to small molecule targets and enzyme design tasks, again surpassing prior methods. Code, models and new data will be publicly released.
Compositional Flows for 3D Molecule and Synthesis Pathway Co-design
Apr 10, 2025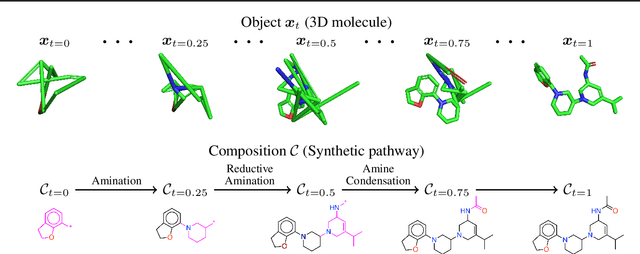
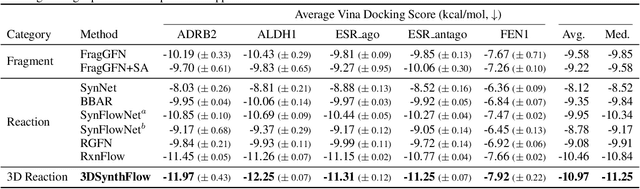
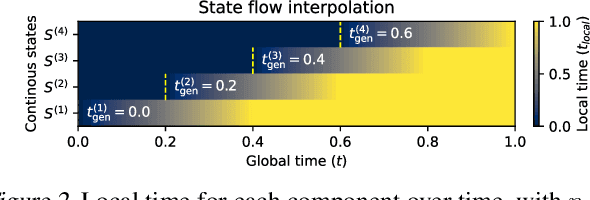
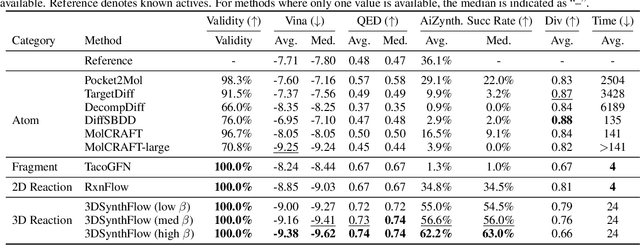
Many generative applications, such as synthesis-based 3D molecular design, involve constructing compositional objects with continuous features. Here, we introduce Compositional Generative Flows (CGFlow), a novel framework that extends flow matching to generate objects in compositional steps while modeling continuous states. Our key insight is that modeling compositional state transitions can be formulated as a straightforward extension of the flow matching interpolation process. We further build upon the theoretical foundations of generative flow networks (GFlowNets), enabling reward-guided sampling of compositional structures. We apply CGFlow to synthesizable drug design by jointly designing the molecule's synthetic pathway with its 3D binding pose. Our approach achieves state-of-the-art binding affinity on all 15 targets from the LIT-PCBA benchmark, and 5.8$\times$ improvement in sampling efficiency compared to 2D synthesis-based baseline. To our best knowledge, our method is also the first to achieve state of-art-performance in both Vina Dock (-9.38) and AiZynth success rate (62.2\%) on the CrossDocked benchmark.
MotifBench: A standardized protein design benchmark for motif-scaffolding problems
Feb 19, 2025The motif-scaffolding problem is a central task in computational protein design: Given the coordinates of atoms in a geometry chosen to confer a desired biochemical function (a motif), the task is to identify diverse protein structures (scaffolds) that include the motif and maintain its geometry. Significant recent progress on motif-scaffolding has been made due to computational evaluation with reliable protein structure prediction and fixed-backbone sequence design methods. However, significant variability in evaluation strategies across publications has hindered comparability of results, challenged reproducibility, and impeded robust progress. In response we introduce MotifBench, comprising (1) a precisely specified pipeline and evaluation metrics, (2) a collection of 30 benchmark problems, and (3) an implementation of this benchmark and leaderboard at github.com/blt2114/MotifBench. The MotifBench test cases are more difficult compared to earlier benchmarks, and include protein design problems for which solutions are known but on which, to the best of our knowledge, state-of-the-art methods fail to identify any solution.
BioNeMo Framework: a modular, high-performance library for AI model development in drug discovery
Nov 15, 2024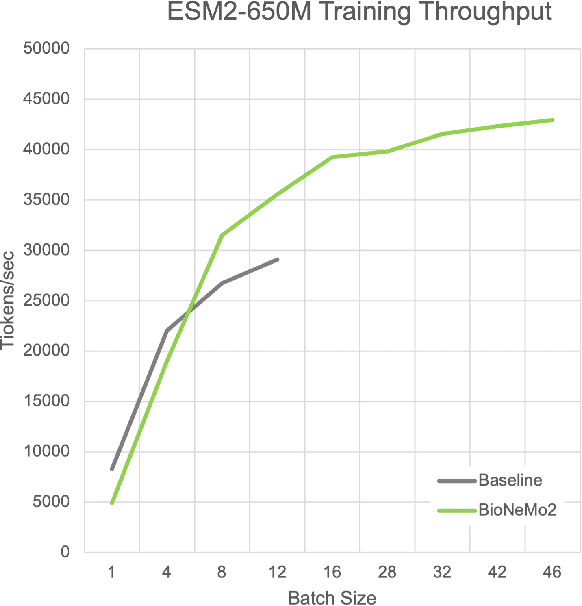
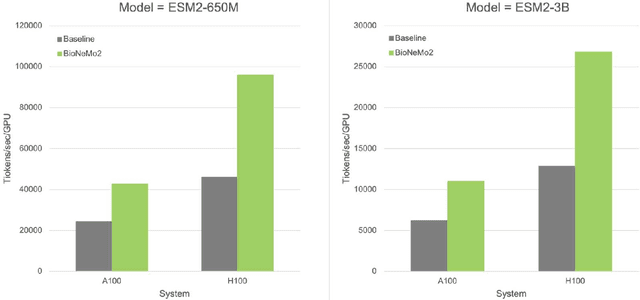
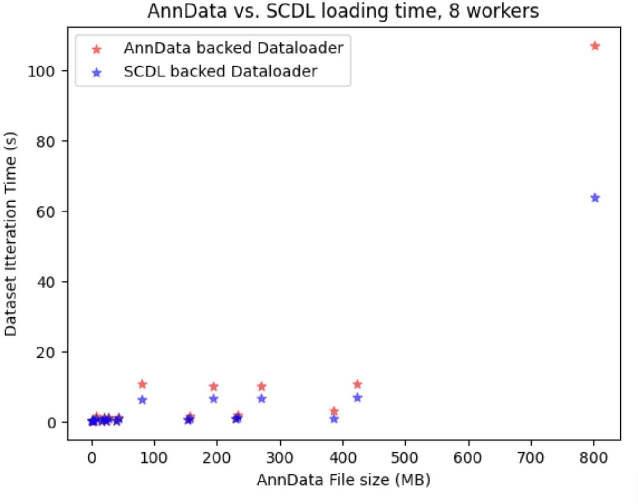
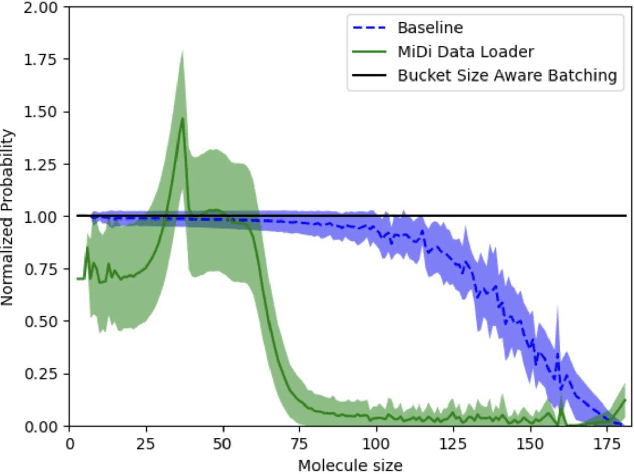
Artificial Intelligence models encoding biology and chemistry are opening new routes to high-throughput and high-quality in-silico drug development. However, their training increasingly relies on computational scale, with recent protein language models (pLM) training on hundreds of graphical processing units (GPUs). We introduce the BioNeMo Framework to facilitate the training of computational biology and chemistry AI models across hundreds of GPUs. Its modular design allows the integration of individual components, such as data loaders, into existing workflows and is open to community contributions. We detail technical features of the BioNeMo Framework through use cases such as pLM pre-training and fine-tuning. On 256 NVIDIA A100s, BioNeMo Framework trains a three billion parameter BERT-based pLM on over one trillion tokens in 4.2 days. The BioNeMo Framework is open-source and free for everyone to use.
RNA-FrameFlow: Flow Matching for de novo 3D RNA Backbone Design
Jun 19, 2024We introduce RNA-FrameFlow, the first generative model for 3D RNA backbone design. We build upon SE(3) flow matching for protein backbone generation and establish protocols for data preparation and evaluation to address unique challenges posed by RNA modeling. We formulate RNA structures as a set of rigid-body frames and associated loss functions which account for larger, more conformationally flexible RNA backbones (13 atoms per nucleotide) vs. proteins (4 atoms per residue). Toward tackling the lack of diversity in 3D RNA datasets, we explore training with structural clustering and cropping augmentations. Additionally, we define a suite of evaluation metrics to measure whether the generated RNA structures are globally self-consistent (via inverse folding followed by forward folding) and locally recover RNA-specific structural descriptors. The most performant version of RNA-FrameFlow generates locally realistic RNA backbones of 40-150 nucleotides, over 40% of which pass our validity criteria as measured by a self-consistency TM-score >= 0.45, at which two RNAs have the same global fold. Open-source code: https://github.com/rish-16/rna-backbone-design
Evaluating representation learning on the protein structure universe
Jun 19, 2024We introduce ProteinWorkshop, a comprehensive benchmark suite for representation learning on protein structures with Geometric Graph Neural Networks. We consider large-scale pre-training and downstream tasks on both experimental and predicted structures to enable the systematic evaluation of the quality of the learned structural representation and their usefulness in capturing functional relationships for downstream tasks. We find that: (1) large-scale pretraining on AlphaFold structures and auxiliary tasks consistently improve the performance of both rotation-invariant and equivariant GNNs, and (2) more expressive equivariant GNNs benefit from pretraining to a greater extent compared to invariant models. We aim to establish a common ground for the machine learning and computational biology communities to rigorously compare and advance protein structure representation learning. Our open-source codebase reduces the barrier to entry for working with large protein structure datasets by providing: (1) storage-efficient dataloaders for large-scale structural databases including AlphaFoldDB and ESM Atlas, as well as (2) utilities for constructing new tasks from the entire PDB. ProteinWorkshop is available at: github.com/a-r-j/ProteinWorkshop.
DEFT: Efficient Finetuning of Conditional Diffusion Models by Learning the Generalised $h$-transform
Jun 03, 2024Generative modelling paradigms based on denoising diffusion processes have emerged as a leading candidate for conditional sampling in inverse problems. In many real-world applications, we often have access to large, expensively trained unconditional diffusion models, which we aim to exploit for improving conditional sampling. Most recent approaches are motivated heuristically and lack a unifying framework, obscuring connections between them. Further, they often suffer from issues such as being very sensitive to hyperparameters, being expensive to train or needing access to weights hidden behind a closed API. In this work, we unify conditional training and sampling using the mathematically well-understood Doob's h-transform. This new perspective allows us to unify many existing methods under a common umbrella. Under this framework, we propose DEFT (Doob's h-transform Efficient FineTuning), a new approach for conditional generation that simply fine-tunes a very small network to quickly learn the conditional $h$-transform, while keeping the larger unconditional network unchanged. DEFT is much faster than existing baselines while achieving state-of-the-art performance across a variety of linear and non-linear benchmarks. On image reconstruction tasks, we achieve speedups of up to 1.6$\times$, while having the best perceptual quality on natural images and reconstruction performance on medical images.
A framework for conditional diffusion modelling with applications in motif scaffolding for protein design
Dec 14, 2023Many protein design applications, such as binder or enzyme design, require scaffolding a structural motif with high precision. Generative modelling paradigms based on denoising diffusion processes emerged as a leading candidate to address this motif scaffolding problem and have shown early experimental success in some cases. In the diffusion paradigm, motif scaffolding is treated as a conditional generation task, and several conditional generation protocols were proposed or imported from the Computer Vision literature. However, most of these protocols are motivated heuristically, e.g. via analogies to Langevin dynamics, and lack a unifying framework, obscuring connections between the different approaches. In this work, we unify conditional training and conditional sampling procedures under one common framework based on the mathematically well-understood Doob's h-transform. This new perspective allows us to draw connections between existing methods and propose a new variation on existing conditional training protocols. We illustrate the effectiveness of this new protocol in both, image outpainting and motif scaffolding and find that it outperforms standard methods.
On How AI Needs to Change to Advance the Science of Drug Discovery
Dec 23, 2022


Research around AI for Science has seen significant success since the rise of deep learning models over the past decade, even with longstanding challenges such as protein structure prediction. However, this fast development inevitably made their flaws apparent -- especially in domains of reasoning where understanding the cause-effect relationship is important. One such domain is drug discovery, in which such understanding is required to make sense of data otherwise plagued by spurious correlations. Said spuriousness only becomes worse with the ongoing trend of ever-increasing amounts of data in the life sciences and thereby restricts researchers in their ability to understand disease biology and create better therapeutics. Therefore, to advance the science of drug discovery with AI it is becoming necessary to formulate the key problems in the language of causality, which allows the explication of modelling assumptions needed for identifying true cause-effect relationships. In this attention paper, we present causal drug discovery as the craft of creating models that ground the process of drug discovery in causal reasoning.