 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDEFT: Efficient Finetuning of Conditional Diffusion Models by Learning the Generalised $h$-transform
Jun 03, 2024Generative modelling paradigms based on denoising diffusion processes have emerged as a leading candidate for conditional sampling in inverse problems. In many real-world applications, we often have access to large, expensively trained unconditional diffusion models, which we aim to exploit for improving conditional sampling. Most recent approaches are motivated heuristically and lack a unifying framework, obscuring connections between them. Further, they often suffer from issues such as being very sensitive to hyperparameters, being expensive to train or needing access to weights hidden behind a closed API. In this work, we unify conditional training and sampling using the mathematically well-understood Doob's h-transform. This new perspective allows us to unify many existing methods under a common umbrella. Under this framework, we propose DEFT (Doob's h-transform Efficient FineTuning), a new approach for conditional generation that simply fine-tunes a very small network to quickly learn the conditional $h$-transform, while keeping the larger unconditional network unchanged. DEFT is much faster than existing baselines while achieving state-of-the-art performance across a variety of linear and non-linear benchmarks. On image reconstruction tasks, we achieve speedups of up to 1.6$\times$, while having the best perceptual quality on natural images and reconstruction performance on medical images.
Predicting protein variants with equivariant graph neural networks
Jun 21, 2023

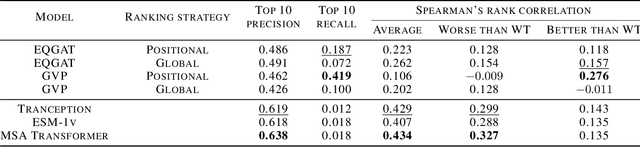

Pre-trained models have been successful in many protein engineering tasks. Most notably, sequence-based models have achieved state-of-the-art performance on protein fitness prediction while structure-based models have been used experimentally to develop proteins with enhanced functions. However, there is a research gap in comparing structure- and sequence-based methods for predicting protein variants that are better than the wildtype protein. This paper aims to address this gap by conducting a comparative study between the abilities of equivariant graph neural networks (EGNNs) and sequence-based approaches to identify promising amino-acid mutations. The results show that our proposed structural approach achieves a competitive performance to sequence-based methods while being trained on significantly fewer molecules. Additionally, we find that combining assay labelled data with structure pre-trained models yields similar trends as with sequence pre-trained models.
Multi-State RNA Design with Geometric Multi-Graph Neural Networks
May 25, 2023Computational RNA design has broad applications across synthetic biology and therapeutic development. Fundamental to the diverse biological functions of RNA is its conformational flexibility, enabling single sequences to adopt a variety of distinct 3D states. Currently, computational biomolecule design tasks are often posed as inverse problems, where sequences are designed based on adopting a single desired structural conformation. In this work, we propose gRNAde, a geometric RNA design pipeline that operates on sets of 3D RNA backbone structures to explicitly account for and reflect RNA conformational diversity in its designs. We demonstrate the utility of gRNAde for improving native sequence recovery over single-state approaches on a new large-scale 3D RNA design dataset, especially for multi-state and structurally diverse RNAs. Our code is available at https://github.com/chaitjo/geometric-rna-design