 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePredicting protein variants with equivariant graph neural networks
Jun 21, 2023

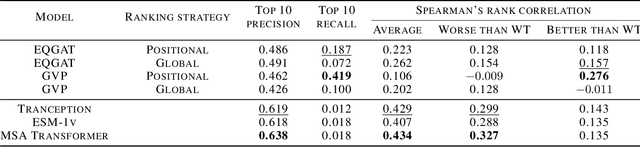

Pre-trained models have been successful in many protein engineering tasks. Most notably, sequence-based models have achieved state-of-the-art performance on protein fitness prediction while structure-based models have been used experimentally to develop proteins with enhanced functions. However, there is a research gap in comparing structure- and sequence-based methods for predicting protein variants that are better than the wildtype protein. This paper aims to address this gap by conducting a comparative study between the abilities of equivariant graph neural networks (EGNNs) and sequence-based approaches to identify promising amino-acid mutations. The results show that our proposed structural approach achieves a competitive performance to sequence-based methods while being trained on significantly fewer molecules. Additionally, we find that combining assay labelled data with structure pre-trained models yields similar trends as with sequence pre-trained models.