 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTABASCO: A Fast, Simplified Model for Molecular Generation with Improved Physical Quality
Jul 01, 2025State-of-the-art models for 3D molecular generation are based on significant inductive biases, SE(3), permutation equivariance to respect symmetry and graph message-passing networks to capture local chemistry, yet the generated molecules still struggle with physical plausibility. We introduce TABASCO which relaxes these assumptions: The model has a standard non-equivariant transformer architecture, treats atoms in a molecule as sequences and reconstructs bonds deterministically after generation. The absence of equivariant layers and message passing allows us to significantly simplify the model architecture and scale data throughput. On the GEOM-Drugs benchmark TABASCO achieves state-of-the-art PoseBusters validity and delivers inference roughly 10x faster than the strongest baseline, while exhibiting emergent rotational equivariance despite symmetry not being hard-coded. Our work offers a blueprint for training minimalist, high-throughput generative models suited to specialised tasks such as structure- and pharmacophore-based drug design. We provide a link to our implementation at github.com/carlosinator/tabasco.
RNA-FrameFlow: Flow Matching for de novo 3D RNA Backbone Design
Jun 19, 2024We introduce RNA-FrameFlow, the first generative model for 3D RNA backbone design. We build upon SE(3) flow matching for protein backbone generation and establish protocols for data preparation and evaluation to address unique challenges posed by RNA modeling. We formulate RNA structures as a set of rigid-body frames and associated loss functions which account for larger, more conformationally flexible RNA backbones (13 atoms per nucleotide) vs. proteins (4 atoms per residue). Toward tackling the lack of diversity in 3D RNA datasets, we explore training with structural clustering and cropping augmentations. Additionally, we define a suite of evaluation metrics to measure whether the generated RNA structures are globally self-consistent (via inverse folding followed by forward folding) and locally recover RNA-specific structural descriptors. The most performant version of RNA-FrameFlow generates locally realistic RNA backbones of 40-150 nucleotides, over 40% of which pass our validity criteria as measured by a self-consistency TM-score >= 0.45, at which two RNAs have the same global fold. Open-source code: https://github.com/rish-16/rna-backbone-design
Evaluating representation learning on the protein structure universe
Jun 19, 2024We introduce ProteinWorkshop, a comprehensive benchmark suite for representation learning on protein structures with Geometric Graph Neural Networks. We consider large-scale pre-training and downstream tasks on both experimental and predicted structures to enable the systematic evaluation of the quality of the learned structural representation and their usefulness in capturing functional relationships for downstream tasks. We find that: (1) large-scale pretraining on AlphaFold structures and auxiliary tasks consistently improve the performance of both rotation-invariant and equivariant GNNs, and (2) more expressive equivariant GNNs benefit from pretraining to a greater extent compared to invariant models. We aim to establish a common ground for the machine learning and computational biology communities to rigorously compare and advance protein structure representation learning. Our open-source codebase reduces the barrier to entry for working with large protein structure datasets by providing: (1) storage-efficient dataloaders for large-scale structural databases including AlphaFoldDB and ESM Atlas, as well as (2) utilities for constructing new tasks from the entire PDB. ProteinWorkshop is available at: github.com/a-r-j/ProteinWorkshop.
SynFlowNet: Towards Molecule Design with Guaranteed Synthesis Pathways
May 02, 2024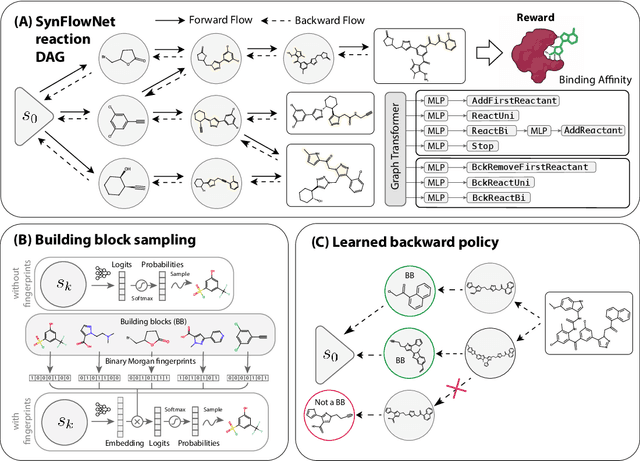

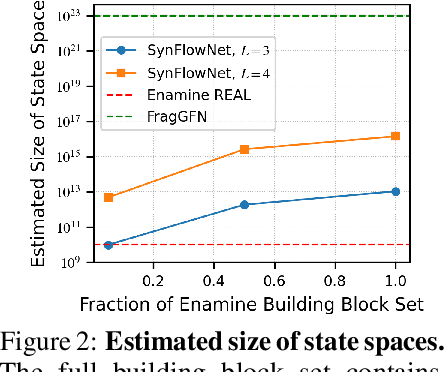
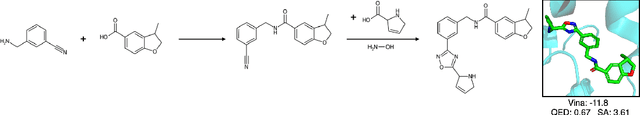
Recent breakthroughs in generative modelling have led to a number of works proposing molecular generation models for drug discovery. While these models perform well at capturing drug-like motifs, they are known to often produce synthetically inaccessible molecules. This is because they are trained to compose atoms or fragments in a way that approximates the training distribution, but they are not explicitly aware of the synthesis constraints that come with making molecules in the lab. To address this issue, we introduce SynFlowNet, a GFlowNet model whose action space uses chemically validated reactions and reactants to sequentially build new molecules. We evaluate our approach using synthetic accessibility scores and an independent retrosynthesis tool. SynFlowNet consistently samples synthetically feasible molecules, while still being able to find diverse and high-utility candidates. Furthermore, we compare molecules designed with SynFlowNet to experimentally validated actives, and find that they show comparable properties of interest, such as molecular weight, SA score and predicted protein binding affinity.
Multi-State RNA Design with Geometric Multi-Graph Neural Networks
May 25, 2023Computational RNA design has broad applications across synthetic biology and therapeutic development. Fundamental to the diverse biological functions of RNA is its conformational flexibility, enabling single sequences to adopt a variety of distinct 3D states. Currently, computational biomolecule design tasks are often posed as inverse problems, where sequences are designed based on adopting a single desired structural conformation. In this work, we propose gRNAde, a geometric RNA design pipeline that operates on sets of 3D RNA backbone structures to explicitly account for and reflect RNA conformational diversity in its designs. We demonstrate the utility of gRNAde for improving native sequence recovery over single-state approaches on a new large-scale 3D RNA design dataset, especially for multi-state and structurally diverse RNAs. Our code is available at https://github.com/chaitjo/geometric-rna-design
Structure-based Drug Design with Equivariant Diffusion Models
Oct 24, 2022Structure-based drug design (SBDD) aims to design small-molecule ligands that bind with high affinity and specificity to pre-determined protein targets. Traditional SBDD pipelines start with large-scale docking of compound libraries from public databases, thus limiting the exploration of chemical space to existent previously studied regions. Recent machine learning methods approached this problem using an atom-by-atom generation approach, which is computationally expensive. In this paper, we formulate SBDD as a 3D-conditional generation problem and present DiffSBDD, an E(3)-equivariant 3D-conditional diffusion model that generates novel ligands conditioned on protein pockets. Furthermore, we curate a new dataset of experimentally determined binding complex data from Binding MOAD to provide a realistic binding scenario that complements the synthetic CrossDocked dataset. Comprehensive in silico experiments demonstrate the efficiency of DiffSBDD in generating novel and diverse drug-like ligands that engage protein pockets with high binding energies as predicted by in silico docking.