 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeZatom-1: A Multimodal Flow Foundation Model for 3D Molecules and Materials
Feb 24, 2026General-purpose 3D chemical modeling encompasses molecules and materials, requiring both generative and predictive capabilities. However, most existing AI approaches are optimized for a single domain (molecules or materials) and a single task (generation or prediction), which limits representation sharing and transfer. We introduce Zatom-1, the first foundation model that unifies generative and predictive learning of 3D molecules and materials. Zatom-1 is a Transformer trained with a multimodal flow matching objective that jointly models discrete atom types and continuous 3D geometries. This approach supports scalable pretraining with predictable gains as model capacity increases, while enabling fast and stable sampling. We use joint generative pretraining as a universal initialization for downstream multi-task prediction of properties, energies, and forces. Empirically, Zatom-1 matches or outperforms specialized baselines on both generative and predictive benchmarks, while reducing the generative inference time by more than an order of magnitude. Our experiments demonstrate positive predictive transfer between chemical domains from joint generative pretraining: modeling materials during pretraining improves molecular property prediction accuracy.
Topotein: Topological Deep Learning for Protein Representation Learning
Sep 04, 2025Protein representation learning (PRL) is crucial for understanding structure-function relationships, yet current sequence- and graph-based methods fail to capture the hierarchical organization inherent in protein structures. We introduce Topotein, a comprehensive framework that applies topological deep learning to PRL through the novel Protein Combinatorial Complex (PCC) and Topology-Complete Perceptron Network (TCPNet). Our PCC represents proteins at multiple hierarchical levels -- from residues to secondary structures to complete proteins -- while preserving geometric information at each level. TCPNet employs SE(3)-equivariant message passing across these hierarchical structures, enabling more effective capture of multi-scale structural patterns. Through extensive experiments on four PRL tasks, TCPNet consistently outperforms state-of-the-art geometric graph neural networks. Our approach demonstrates particular strength in tasks such as fold classification which require understanding of secondary structure arrangements, validating the importance of hierarchical topological features for protein analysis.
FlowDock: Geometric Flow Matching for Generative Protein-Ligand Docking and Affinity Prediction
Dec 14, 2024



Powerful generative models of protein-ligand structure have recently been proposed, but few of these methods support both flexible protein-ligand docking and affinity estimation. Of those that do, none can directly model multiple binding ligands concurrently or have been rigorously benchmarked on pharmacologically relevant drug targets, hindering their widespread adoption in drug discovery efforts. In this work, we propose FlowDock, a deep geometric generative model based on conditional flow matching that learns to directly map unbound (apo) structures to their bound (holo) counterparts for an arbitrary number of binding ligands. Furthermore, FlowDock provides predicted structural confidence scores and binding affinity values with each of its generated protein-ligand complex structures, enabling fast virtual screening of new (multi-ligand) drug targets. For the commonly-used PoseBusters Benchmark dataset, FlowDock achieves a 51% blind docking success rate using unbound (apo) protein input structures and without any information derived from multiple sequence alignments, and for the challenging new DockGen-E dataset, FlowDock matches the performance of single-sequence Chai-1 for binding pocket generalization. Additionally, in the ligand category of the 16th community-wide Critical Assessment of Techniques for Structure Prediction (CASP16), FlowDock ranked among the top-5 methods for pharmacological binding affinity estimation across 140 protein-ligand complexes, demonstrating the efficacy of its learned representations in virtual screening. Source code, data, and pre-trained models are available at https://github.com/BioinfoMachineLearning/FlowDock.
Evaluating representation learning on the protein structure universe
Jun 19, 2024We introduce ProteinWorkshop, a comprehensive benchmark suite for representation learning on protein structures with Geometric Graph Neural Networks. We consider large-scale pre-training and downstream tasks on both experimental and predicted structures to enable the systematic evaluation of the quality of the learned structural representation and their usefulness in capturing functional relationships for downstream tasks. We find that: (1) large-scale pretraining on AlphaFold structures and auxiliary tasks consistently improve the performance of both rotation-invariant and equivariant GNNs, and (2) more expressive equivariant GNNs benefit from pretraining to a greater extent compared to invariant models. We aim to establish a common ground for the machine learning and computational biology communities to rigorously compare and advance protein structure representation learning. Our open-source codebase reduces the barrier to entry for working with large protein structure datasets by providing: (1) storage-efficient dataloaders for large-scale structural databases including AlphaFoldDB and ESM Atlas, as well as (2) utilities for constructing new tasks from the entire PDB. ProteinWorkshop is available at: github.com/a-r-j/ProteinWorkshop.
RNA-FrameFlow: Flow Matching for de novo 3D RNA Backbone Design
Jun 19, 2024We introduce RNA-FrameFlow, the first generative model for 3D RNA backbone design. We build upon SE(3) flow matching for protein backbone generation and establish protocols for data preparation and evaluation to address unique challenges posed by RNA modeling. We formulate RNA structures as a set of rigid-body frames and associated loss functions which account for larger, more conformationally flexible RNA backbones (13 atoms per nucleotide) vs. proteins (4 atoms per residue). Toward tackling the lack of diversity in 3D RNA datasets, we explore training with structural clustering and cropping augmentations. Additionally, we define a suite of evaluation metrics to measure whether the generated RNA structures are globally self-consistent (via inverse folding followed by forward folding) and locally recover RNA-specific structural descriptors. The most performant version of RNA-FrameFlow generates locally realistic RNA backbones of 40-150 nucleotides, over 40% of which pass our validity criteria as measured by a self-consistency TM-score >= 0.45, at which two RNAs have the same global fold. Open-source code: https://github.com/rish-16/rna-backbone-design
Deep Learning for Protein-Ligand Docking: Are We There Yet?
May 23, 2024



The effects of ligand binding on protein structures and their in vivo functions carry numerous implications for modern biomedical research and biotechnology development efforts such as drug discovery. Although several deep learning (DL) methods and benchmarks designed for protein-ligand docking have recently been introduced, to date no prior works have systematically studied the behavior of docking methods within the practical context of (1) predicted (apo) protein structures, (2) multiple ligands concurrently binding to a given target protein, and (3) having no prior knowledge of binding pockets. To enable a deeper understanding of docking methods' real-world utility, we introduce PoseBench, the first comprehensive benchmark for practical protein-ligand docking. PoseBench enables researchers to rigorously and systematically evaluate DL docking methods for apo-to-holo protein-ligand docking and protein-ligand structure generation using both single and multi-ligand benchmark datasets, the latter of which we introduce for the first time to the DL community. Empirically, using PoseBench, we find that all recent DL docking methods but one fail to generalize to multi-ligand protein targets and also that template-based docking algorithms perform equally well or better for multi-ligand docking as recent single-ligand DL docking methods, suggesting areas of improvement for future work. Code, data, tutorials, and benchmark results are available at https://github.com/BioinfoMachineLearning/PoseBench.
Towards Joint Sequence-Structure Generation of Nucleic Acid and Protein Complexes with SE(3)-Discrete Diffusion
Dec 21, 2023Generative models of macromolecules carry abundant and impactful implications for industrial and biomedical efforts in protein engineering. However, existing methods are currently limited to modeling protein structures or sequences, independently or jointly, without regard to the interactions that commonly occur between proteins and other macromolecules. In this work, we introduce MMDiff, a generative model that jointly designs sequences and structures of nucleic acid and protein complexes, independently or in complex, using joint SE(3)-discrete diffusion noise. Such a model has important implications for emerging areas of macromolecular design including structure-based transcription factor design and design of noncoding RNA sequences. We demonstrate the utility of MMDiff through a rigorous new design benchmark for macromolecular complex generation that we introduce in this work. Our results demonstrate that MMDiff is able to successfully generate micro-RNA and single-stranded DNA molecules while being modestly capable of joint modeling DNA and RNA molecules in interaction with multi-chain protein complexes. Source code: https://github.com/Profluent-Internships/MMDiff.
Geometry-Complete Diffusion for 3D Molecule Generation
Feb 15, 2023Denoising diffusion probabilistic models (DDPMs) have recently taken the field of generative modeling by storm, pioneering new state-of-the-art results in disciplines such as computer vision and computational biology for diverse tasks ranging from text-guided image generation to structure-guided protein design. Along this latter line of research, methods such as those of Hoogeboom et al. 2022 have been proposed for unconditionally generating 3D molecules using equivariant graph neural networks (GNNs) within a DDPM framework. Toward this end, we propose GCDM, a geometry-complete diffusion model that achieves new state-of-the-art results for 3D molecule diffusion generation by leveraging the representation learning strengths offered by GNNs that perform geometry-complete message-passing. Our results with GCDM also offer preliminary insights into how physical inductive biases impact the generative dynamics of molecular DDPMs. The source code, data, and instructions to train new models or reproduce our results are freely available at https://github.com/BioinfoMachineLearning/bio-diffusion.
Geometry-Complete Perceptron Networks for 3D Molecular Graphs
Nov 04, 2022



The field of geometric deep learning has had a profound impact on the development of innovative and powerful graph neural network architectures. Disciplines such as computer vision and computational biology have benefited significantly from such methodological advances, which has led to breakthroughs in scientific domains such as protein structure prediction and design. In this work, we introduce GCPNet, a new geometry-complete, SE(3)-equivariant graph neural network designed for 3D graph representation learning. We demonstrate the state-of-the-art utility and expressiveness of our method on six independent datasets designed for three distinct geometric tasks: protein-ligand binding affinity prediction, protein structure ranking, and Newtonian many-body systems modeling. Our results suggest that GCPNet is a powerful, general method for capturing complex geometric and physical interactions within 3D graphs for downstream prediction tasks. The source code, data, and instructions to train new models or reproduce our results are freely available on GitHub.
DRLComplex: Reconstruction of protein quaternary structures using deep reinforcement learning
May 26, 2022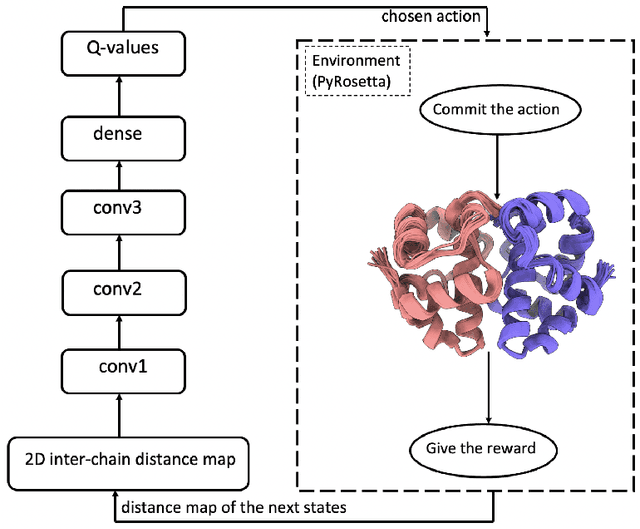
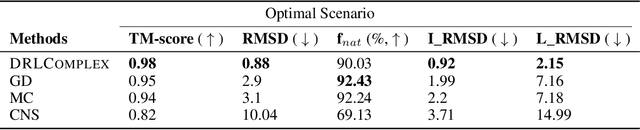
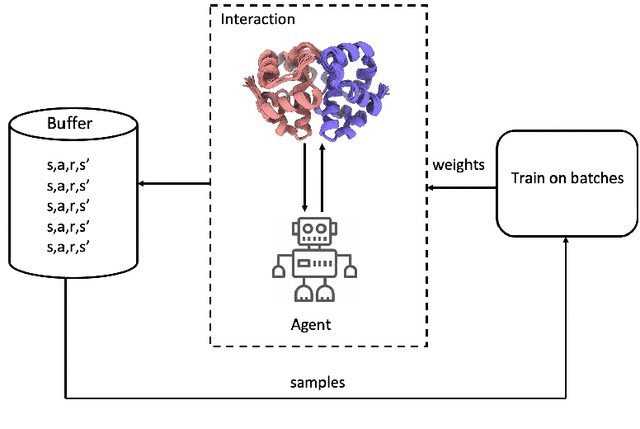

Predicted inter-chain residue-residue contacts can be used to build the quaternary structure of protein complexes from scratch. However, only a small number of methods have been developed to reconstruct protein quaternary structures using predicted inter-chain contacts. Here, we present an agent-based self-learning method based on deep reinforcement learning (DRLComplex) to build protein complex structures using inter-chain contacts as distance constraints. We rigorously tested DRLComplex on two standard datasets of homodimeric and heterodimeric protein complexes (i.e., the CASP-CAPRI homodimer and Std_32 heterodimer datasets) using both true and predicted interchain contacts as inputs. Utilizing true contacts as input, DRLComplex achieved high average TM-scores of 0.9895 and 0.9881 and a low average interface RMSD (I_RMSD) of 0.2197 and 0.92 on the two datasets, respectively. When predicted contacts are used, the method achieves TM-scores of 0.73 and 0.76 for homodimers and heterodimers, respectively. Our experiments find that the accuracy of reconstructed quaternary structures depends on the accuracy of the contact predictions. Compared to other optimization methods for reconstructing quaternary structures from inter-chain contacts, DRLComplex performs similar to an advanced gradient descent method and better than a Markov Chain Monte Carlo simulation method and a simulated annealing-based method, validating the effectiveness of DRLComplex for quaternary reconstruction of protein complexes.