 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHalluMat: Detecting Hallucinations in LLM-Generated Materials Science Content Through Multi-Stage Verification
Dec 26, 2025Artificial Intelligence (AI), particularly Large Language Models (LLMs), is transforming scientific discovery, enabling rapid knowledge generation and hypothesis formulation. However, a critical challenge is hallucination, where LLMs generate factually incorrect or misleading information, compromising research integrity. To address this, we introduce HalluMatData, a benchmark dataset for evaluating hallucination detection methods, factual consistency, and response robustness in AI-generated materials science content. Alongside this, we propose HalluMatDetector, a multi-stage hallucination detection framework that integrates intrinsic verification, multi-source retrieval, contradiction graph analysis, and metric-based assessment to detect and mitigate LLM hallucinations. Our findings reveal that hallucination levels vary significantly across materials science subdomains, with high-entropy queries exhibiting greater factual inconsistencies. By utilizing HalluMatDetector verification pipeline, we reduce hallucination rates by 30% compared to standard LLM outputs. Furthermore, we introduce the Paraphrased Hallucination Consistency Score (PHCS) to quantify inconsistencies in LLM responses across semantically equivalent queries, offering deeper insights into model reliability.
Machine Learning Methods for Gene Regulatory Network Inference
Apr 17, 2025



Gene Regulatory Networks (GRNs) are intricate biological systems that control gene expression and regulation in response to environmental and developmental cues. Advances in computational biology, coupled with high throughput sequencing technologies, have significantly improved the accuracy of GRN inference and modeling. Modern approaches increasingly leverage artificial intelligence (AI), particularly machine learning techniques including supervised, unsupervised, semi-supervised, and contrastive learning to analyze large scale omics data and uncover regulatory gene interactions. To support both the application of GRN inference in studying gene regulation and the development of novel machine learning methods, we present a comprehensive review of machine learning based GRN inference methodologies, along with the datasets and evaluation metrics commonly used. Special emphasis is placed on the emerging role of cutting edge deep learning techniques in enhancing inference performance. The potential future directions for improving GRN inference are also discussed.
FlowDock: Geometric Flow Matching for Generative Protein-Ligand Docking and Affinity Prediction
Dec 14, 2024



Powerful generative models of protein-ligand structure have recently been proposed, but few of these methods support both flexible protein-ligand docking and affinity estimation. Of those that do, none can directly model multiple binding ligands concurrently or have been rigorously benchmarked on pharmacologically relevant drug targets, hindering their widespread adoption in drug discovery efforts. In this work, we propose FlowDock, a deep geometric generative model based on conditional flow matching that learns to directly map unbound (apo) structures to their bound (holo) counterparts for an arbitrary number of binding ligands. Furthermore, FlowDock provides predicted structural confidence scores and binding affinity values with each of its generated protein-ligand complex structures, enabling fast virtual screening of new (multi-ligand) drug targets. For the commonly-used PoseBusters Benchmark dataset, FlowDock achieves a 51% blind docking success rate using unbound (apo) protein input structures and without any information derived from multiple sequence alignments, and for the challenging new DockGen-E dataset, FlowDock matches the performance of single-sequence Chai-1 for binding pocket generalization. Additionally, in the ligand category of the 16th community-wide Critical Assessment of Techniques for Structure Prediction (CASP16), FlowDock ranked among the top-5 methods for pharmacological binding affinity estimation across 140 protein-ligand complexes, demonstrating the efficacy of its learned representations in virtual screening. Source code, data, and pre-trained models are available at https://github.com/BioinfoMachineLearning/FlowDock.
Evaluating representation learning on the protein structure universe
Jun 19, 2024We introduce ProteinWorkshop, a comprehensive benchmark suite for representation learning on protein structures with Geometric Graph Neural Networks. We consider large-scale pre-training and downstream tasks on both experimental and predicted structures to enable the systematic evaluation of the quality of the learned structural representation and their usefulness in capturing functional relationships for downstream tasks. We find that: (1) large-scale pretraining on AlphaFold structures and auxiliary tasks consistently improve the performance of both rotation-invariant and equivariant GNNs, and (2) more expressive equivariant GNNs benefit from pretraining to a greater extent compared to invariant models. We aim to establish a common ground for the machine learning and computational biology communities to rigorously compare and advance protein structure representation learning. Our open-source codebase reduces the barrier to entry for working with large protein structure datasets by providing: (1) storage-efficient dataloaders for large-scale structural databases including AlphaFoldDB and ESM Atlas, as well as (2) utilities for constructing new tasks from the entire PDB. ProteinWorkshop is available at: github.com/a-r-j/ProteinWorkshop.
Deep Learning for Protein-Ligand Docking: Are We There Yet?
May 23, 2024



The effects of ligand binding on protein structures and their in vivo functions carry numerous implications for modern biomedical research and biotechnology development efforts such as drug discovery. Although several deep learning (DL) methods and benchmarks designed for protein-ligand docking have recently been introduced, to date no prior works have systematically studied the behavior of docking methods within the practical context of (1) predicted (apo) protein structures, (2) multiple ligands concurrently binding to a given target protein, and (3) having no prior knowledge of binding pockets. To enable a deeper understanding of docking methods' real-world utility, we introduce PoseBench, the first comprehensive benchmark for practical protein-ligand docking. PoseBench enables researchers to rigorously and systematically evaluate DL docking methods for apo-to-holo protein-ligand docking and protein-ligand structure generation using both single and multi-ligand benchmark datasets, the latter of which we introduce for the first time to the DL community. Empirically, using PoseBench, we find that all recent DL docking methods but one fail to generalize to multi-ligand protein targets and also that template-based docking algorithms perform equally well or better for multi-ligand docking as recent single-ligand DL docking methods, suggesting areas of improvement for future work. Code, data, tutorials, and benchmark results are available at https://github.com/BioinfoMachineLearning/PoseBench.
Geometry-Complete Diffusion for 3D Molecule Generation
Feb 15, 2023Denoising diffusion probabilistic models (DDPMs) have recently taken the field of generative modeling by storm, pioneering new state-of-the-art results in disciplines such as computer vision and computational biology for diverse tasks ranging from text-guided image generation to structure-guided protein design. Along this latter line of research, methods such as those of Hoogeboom et al. 2022 have been proposed for unconditionally generating 3D molecules using equivariant graph neural networks (GNNs) within a DDPM framework. Toward this end, we propose GCDM, a geometry-complete diffusion model that achieves new state-of-the-art results for 3D molecule diffusion generation by leveraging the representation learning strengths offered by GNNs that perform geometry-complete message-passing. Our results with GCDM also offer preliminary insights into how physical inductive biases impact the generative dynamics of molecular DDPMs. The source code, data, and instructions to train new models or reproduce our results are freely available at https://github.com/BioinfoMachineLearning/bio-diffusion.
Diffusion Models in Bioinformatics: A New Wave of Deep Learning Revolution in Action
Feb 13, 2023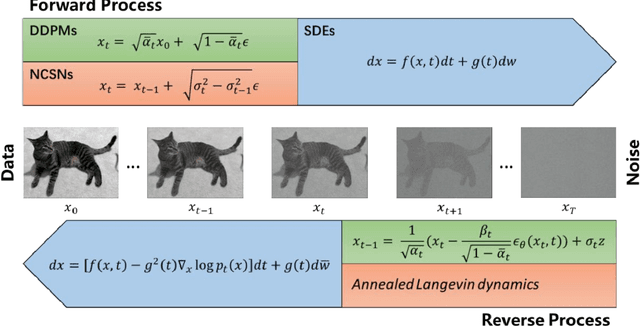
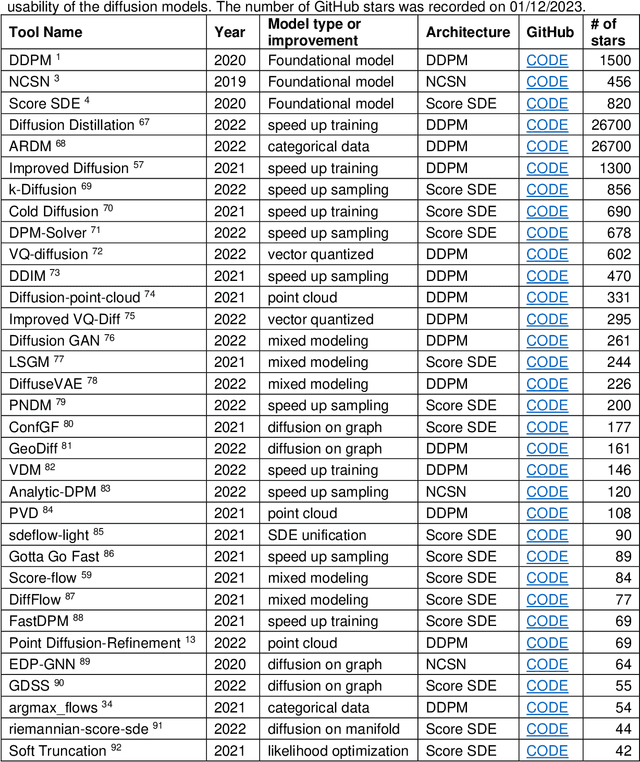
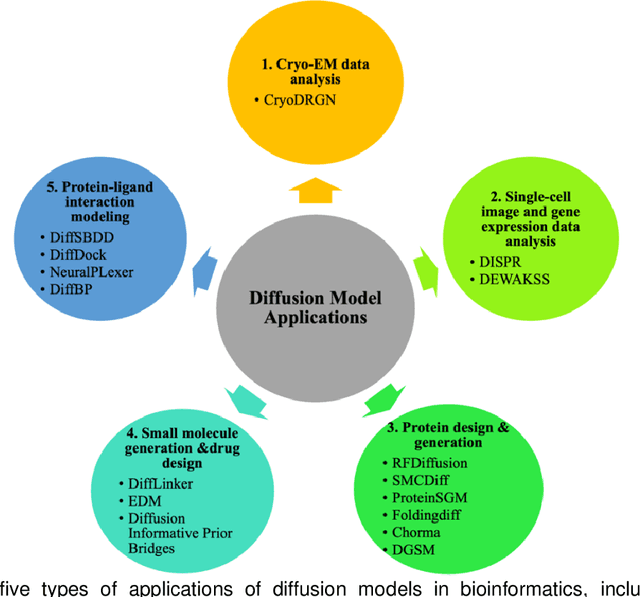
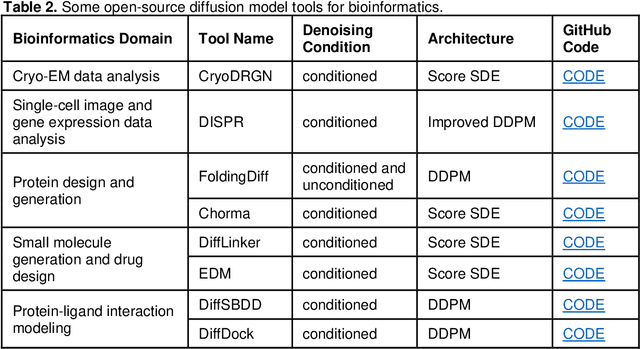
Denoising diffusion models have emerged as one of the most powerful generative models in recent years. They have achieved remarkable success in many fields, such as computer vision, natural language processing (NLP), and bioinformatics. Although there are a few excellent reviews on diffusion models and their applications in computer vision and NLP, there is a lack of an overview of their applications in bioinformatics. This review aims to provide a rather thorough overview of the applications of diffusion models in bioinformatics to aid their further development in bioinformatics and computational biology. We start with an introduction of the key concepts and theoretical foundations of three cornerstone diffusion modeling frameworks (denoising diffusion probabilistic models, noise-conditioned scoring networks, and stochastic differential equations), followed by a comprehensive description of diffusion models employed in the different domains of bioinformatics, including cryo-EM data enhancement, single-cell data analysis, protein design and generation, drug and small molecule design, and protein-ligand interaction. The review is concluded with a summary of the potential new development and applications of diffusion models in bioinformatics.
Geometry-Complete Perceptron Networks for 3D Molecular Graphs
Nov 04, 2022



The field of geometric deep learning has had a profound impact on the development of innovative and powerful graph neural network architectures. Disciplines such as computer vision and computational biology have benefited significantly from such methodological advances, which has led to breakthroughs in scientific domains such as protein structure prediction and design. In this work, we introduce GCPNet, a new geometry-complete, SE(3)-equivariant graph neural network designed for 3D graph representation learning. We demonstrate the state-of-the-art utility and expressiveness of our method on six independent datasets designed for three distinct geometric tasks: protein-ligand binding affinity prediction, protein structure ranking, and Newtonian many-body systems modeling. Our results suggest that GCPNet is a powerful, general method for capturing complex geometric and physical interactions within 3D graphs for downstream prediction tasks. The source code, data, and instructions to train new models or reproduce our results are freely available on GitHub.
Deep learning for reconstructing protein structures from cryo-EM density maps: recent advances and future directions
Sep 16, 2022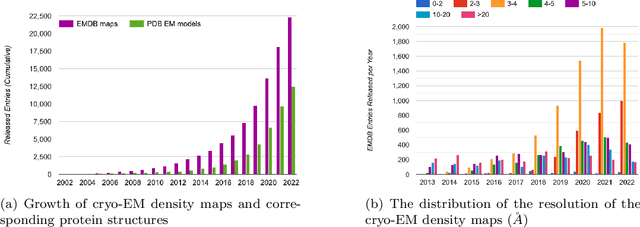

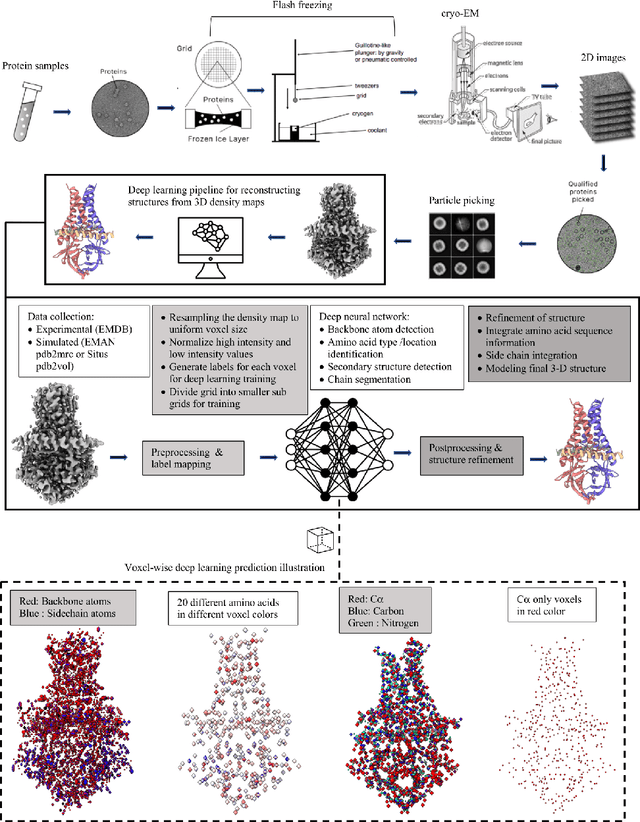
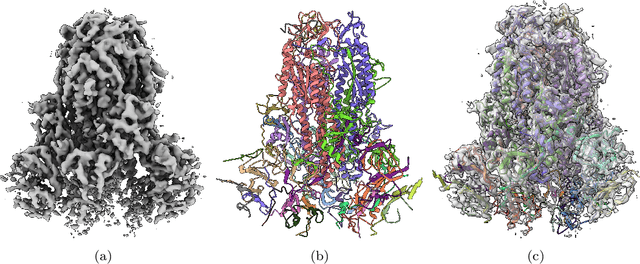
Cryo-Electron Microscopy (cryo-EM) has emerged as a key technology to determine the structure of proteins, particularly large protein complexes and assemblies in recent years. A key challenge in cryo-EM data analysis is to automatically reconstruct accurate protein structures from cryo-EM density maps. In this review, we briefly overview various deep learning methods for building protein structures from cryo-EM density maps, analyze their impact, and discuss the challenges of preparing high-quality data sets for training deep learning models. Looking into the future, more advanced deep learning models of effectively integrating cryo-EM data with other sources of complementary data such as protein sequences and AlphaFold-predicted structures need to be developed to further advance the field.
Deep Learning Prediction of Severe Health Risks for Pediatric COVID-19 Patients with a Large Feature Set in 2021 BARDA Data Challenge
Jun 06, 2022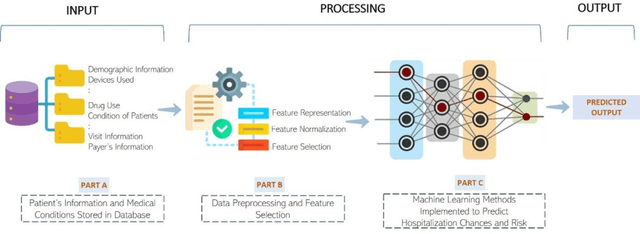

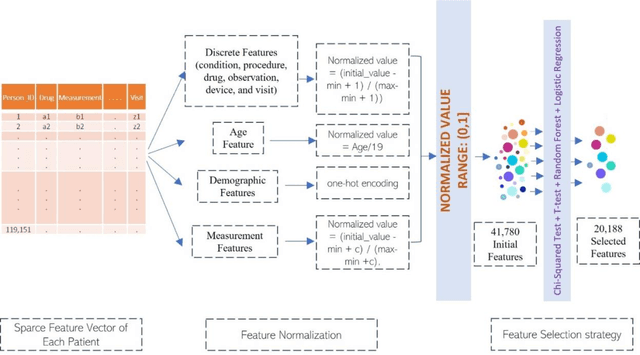
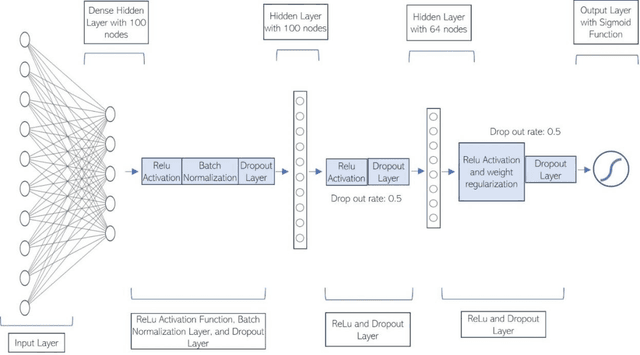
Most children infected with COVID-19 have no or mild symptoms and can recover automatically by themselves, but some pediatric COVID-19 patients need to be hospitalized or even to receive intensive medical care (e.g., invasive mechanical ventilation or cardiovascular support) to recover from the illnesses. Therefore, it is critical to predict the severe health risk that COVID-19 infection poses to children to provide precise and timely medical care for vulnerable pediatric COVID-19 patients. However, predicting the severe health risk for COVID-19 patients including children remains a significant challenge because many underlying medical factors affecting the risk are still largely unknown. In this work, instead of searching for a small number of most useful features to make prediction, we design a novel large-scale bag-of-words like method to represent various medical conditions and measurements of COVID-19 patients. After some simple feature filtering based on logistical regression, the large set of features is used with a deep learning method to predict both the hospitalization risk for COVID-19 infected children and the severe complication risk for the hospitalized pediatric COVID-19 patients. The method was trained and tested the datasets of the Biomedical Advanced Research and Development Authority (BARDA) Pediatric COVID-19 Data Challenge held from Sept. 15 to Dec. 17, 2021. The results show that the approach can rather accurately predict the risk of hospitalization and severe complication for pediatric COVID-19 patients and deep learning is more accurate than other machine learning methods.