 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeYeti: A compact protein structure tokenizer for reconstruction and multi-modal generation
May 11, 2026Multimodal models that jointly reason over protein sequences, structures, and function annotations within a unified representation hold immense potential for integrating multimodal data and generating new proteins with designed functional properties. To utilize transformer architectures, such models require a tokenizer that converts protein structure from continuous atomic coordinates into discrete representations suitable for scalable multimodal training. The quality of such models are fundamentally upper bounded by the fidelity and expressiveness of the underlying tokenized structure. However, existing tokenizers prioritize reconstruction over generative abilities. To address these gaps, we introduce Yeti, a simple and compact protein structure tokenizer based on lookup free quantization and trained end to end with a flow matching objective for multimodal learning. Compared to existing models, Yeti generally achieves the best codebook utilization and token diversity, and second best reconstruction accuracy (with 10x fewer parameters than ESM3) on diverse datasets. To validate Yeti's generative capability, we trained a compact multimodal model jointly over its structure tokens and amino acid sequence entirely from scratch, with no pretrained initialization. The resulting multimodal model generates plausible structures under unconditional cogeneration of protein sequence and structures, achieving comparable results to 10x larger models. Together, these results demonstrate that Yeti is a compact and expressive protein structure tokenizer suitable for training multimodal models that cogenerates highly plausible sequences and structures.
Deep Learning for Protein-Ligand Docking: Are We There Yet?
May 23, 2024



The effects of ligand binding on protein structures and their in vivo functions carry numerous implications for modern biomedical research and biotechnology development efforts such as drug discovery. Although several deep learning (DL) methods and benchmarks designed for protein-ligand docking have recently been introduced, to date no prior works have systematically studied the behavior of docking methods within the practical context of (1) predicted (apo) protein structures, (2) multiple ligands concurrently binding to a given target protein, and (3) having no prior knowledge of binding pockets. To enable a deeper understanding of docking methods' real-world utility, we introduce PoseBench, the first comprehensive benchmark for practical protein-ligand docking. PoseBench enables researchers to rigorously and systematically evaluate DL docking methods for apo-to-holo protein-ligand docking and protein-ligand structure generation using both single and multi-ligand benchmark datasets, the latter of which we introduce for the first time to the DL community. Empirically, using PoseBench, we find that all recent DL docking methods but one fail to generalize to multi-ligand protein targets and also that template-based docking algorithms perform equally well or better for multi-ligand docking as recent single-ligand DL docking methods, suggesting areas of improvement for future work. Code, data, tutorials, and benchmark results are available at https://github.com/BioinfoMachineLearning/PoseBench.
Deep learning for reconstructing protein structures from cryo-EM density maps: recent advances and future directions
Sep 16, 2022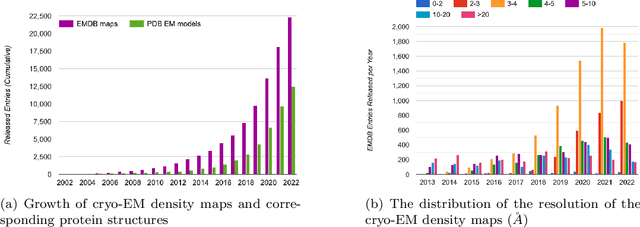

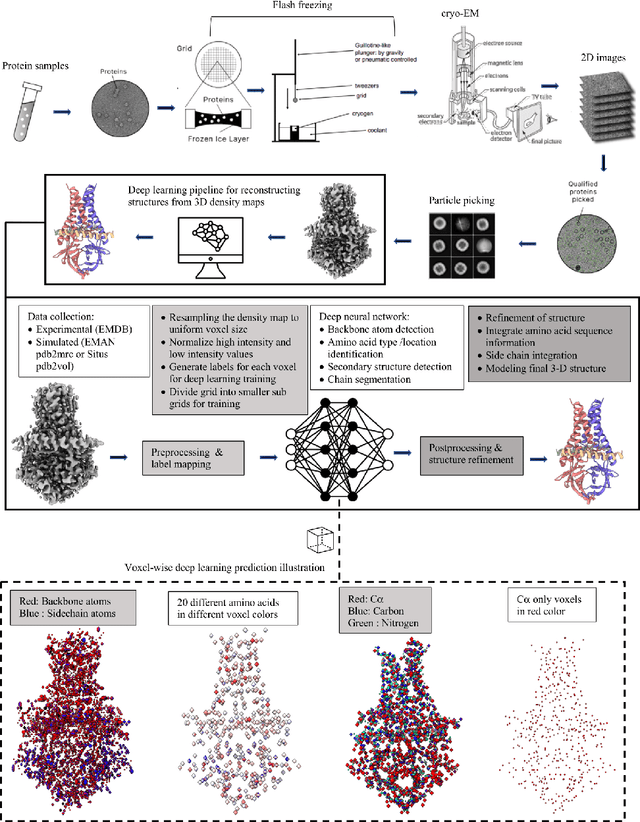
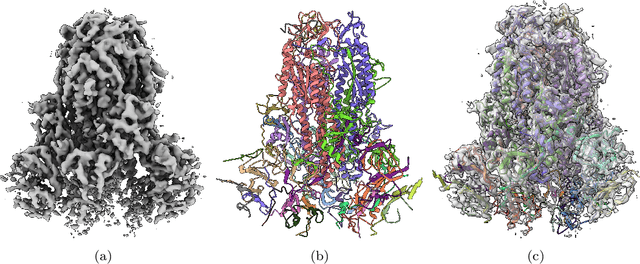
Cryo-Electron Microscopy (cryo-EM) has emerged as a key technology to determine the structure of proteins, particularly large protein complexes and assemblies in recent years. A key challenge in cryo-EM data analysis is to automatically reconstruct accurate protein structures from cryo-EM density maps. In this review, we briefly overview various deep learning methods for building protein structures from cryo-EM density maps, analyze their impact, and discuss the challenges of preparing high-quality data sets for training deep learning models. Looking into the future, more advanced deep learning models of effectively integrating cryo-EM data with other sources of complementary data such as protein sequences and AlphaFold-predicted structures need to be developed to further advance the field.
DRLComplex: Reconstruction of protein quaternary structures using deep reinforcement learning
May 26, 2022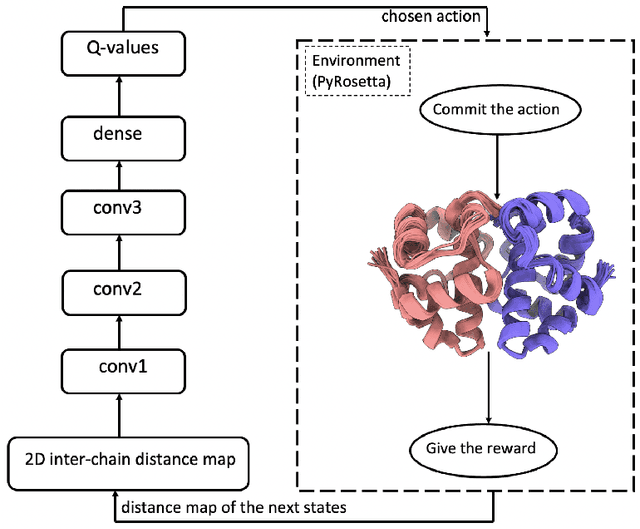
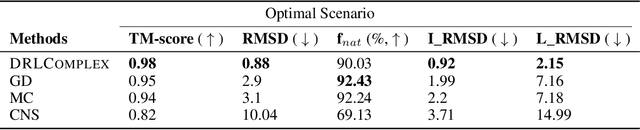

Predicted inter-chain residue-residue contacts can be used to build the quaternary structure of protein complexes from scratch. However, only a small number of methods have been developed to reconstruct protein quaternary structures using predicted inter-chain contacts. Here, we present an agent-based self-learning method based on deep reinforcement learning (DRLComplex) to build protein complex structures using inter-chain contacts as distance constraints. We rigorously tested DRLComplex on two standard datasets of homodimeric and heterodimeric protein complexes (i.e., the CASP-CAPRI homodimer and Std_32 heterodimer datasets) using both true and predicted interchain contacts as inputs. Utilizing true contacts as input, DRLComplex achieved high average TM-scores of 0.9895 and 0.9881 and a low average interface RMSD (I_RMSD) of 0.2197 and 0.92 on the two datasets, respectively. When predicted contacts are used, the method achieves TM-scores of 0.73 and 0.76 for homodimers and heterodimers, respectively. Our experiments find that the accuracy of reconstructed quaternary structures depends on the accuracy of the contact predictions. Compared to other optimization methods for reconstructing quaternary structures from inter-chain contacts, DRLComplex performs similar to an advanced gradient descent method and better than a Markov Chain Monte Carlo simulation method and a simulated annealing-based method, validating the effectiveness of DRLComplex for quaternary reconstruction of protein complexes.