 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeConditional Enzyme Generation Using Protein Language Models with Adapters
Oct 04, 2024



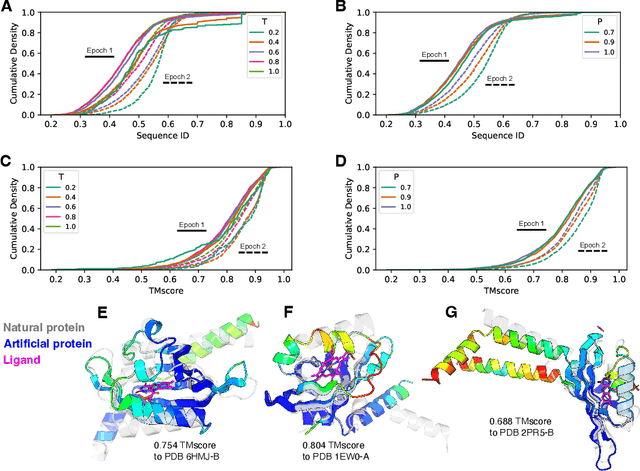
The conditional generation of proteins with desired functions and/or properties is a key goal for generative models. Existing methods based on prompting of language models can generate proteins conditioned on a target functionality, such as a desired enzyme family. However, these methods are limited to simple, tokenized conditioning and have not been shown to generalize to unseen functions. In this study, we propose ProCALM (Protein Conditionally Adapted Language Model), an approach for the conditional generation of proteins using adapters to protein language models. Our specific implementation of ProCALM involves finetuning ProGen2 to incorporate conditioning representations of enzyme function and taxonomy. ProCALM matches existing methods at conditionally generating sequences from target enzyme families. Impressively, it can also generate within the joint distribution of enzymatic function and taxonomy, and it can generalize to rare and unseen enzyme families and taxonomies. Overall, ProCALM is a flexible and computationally efficient approach, and we expect that it can be extended to a wide range of generative language models.
Towards Joint Sequence-Structure Generation of Nucleic Acid and Protein Complexes with SE(3)-Discrete Diffusion
Dec 21, 2023Generative models of macromolecules carry abundant and impactful implications for industrial and biomedical efforts in protein engineering. However, existing methods are currently limited to modeling protein structures or sequences, independently or jointly, without regard to the interactions that commonly occur between proteins and other macromolecules. In this work, we introduce MMDiff, a generative model that jointly designs sequences and structures of nucleic acid and protein complexes, independently or in complex, using joint SE(3)-discrete diffusion noise. Such a model has important implications for emerging areas of macromolecular design including structure-based transcription factor design and design of noncoding RNA sequences. We demonstrate the utility of MMDiff through a rigorous new design benchmark for macromolecular complex generation that we introduce in this work. Our results demonstrate that MMDiff is able to successfully generate micro-RNA and single-stranded DNA molecules while being modestly capable of joint modeling DNA and RNA molecules in interaction with multi-chain protein complexes. Source code: https://github.com/Profluent-Internships/MMDiff.
ProGen2: Exploring the Boundaries of Protein Language Models
Jun 27, 2022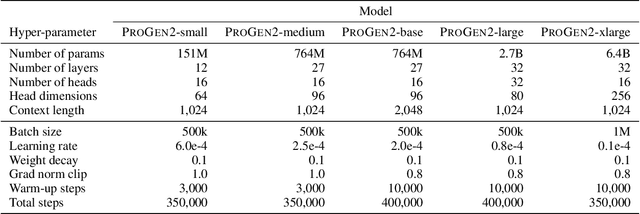
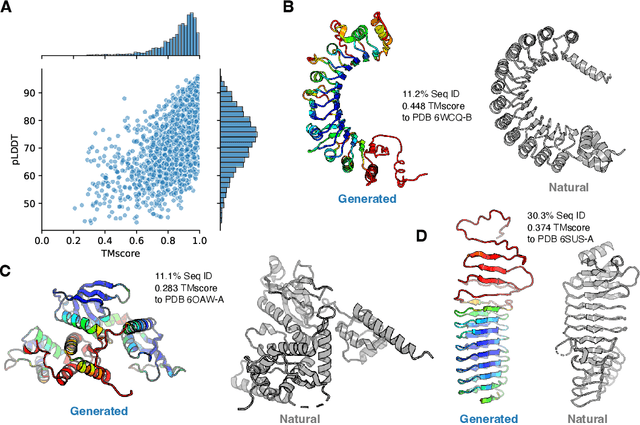
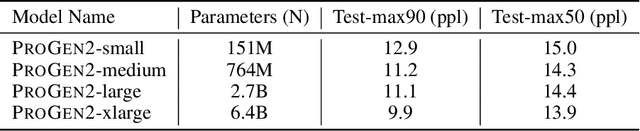
Attention-based models trained on protein sequences have demonstrated incredible success at classification and generation tasks relevant for artificial intelligence-driven protein design. However, we lack a sufficient understanding of how very large-scale models and data play a role in effective protein model development. We introduce a suite of protein language models, named ProGen2, that are scaled up to 6.4B parameters and trained on different sequence datasets drawn from over a billion proteins from genomic, metagenomic, and immune repertoire databases. ProGen2 models show state-of-the-art performance in capturing the distribution of observed evolutionary sequences, generating novel viable sequences, and predicting protein fitness without additional finetuning. As large model sizes and raw numbers of protein sequences continue to become more widely accessible, our results suggest that a growing emphasis needs to be placed on the data distribution provided to a protein sequence model. We release the ProGen2 models and code at https://github.com/salesforce/progen.
Deep Extrapolation for Attribute-Enhanced Generation
Jul 07, 2021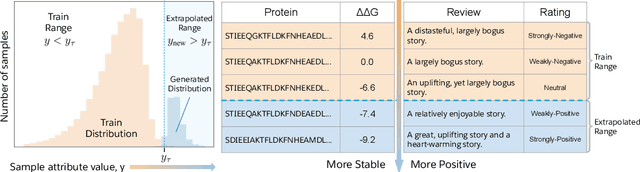
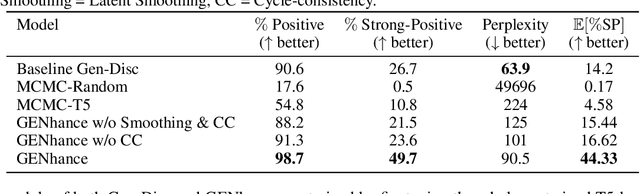
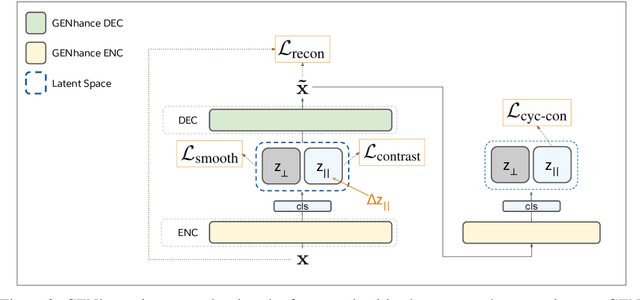
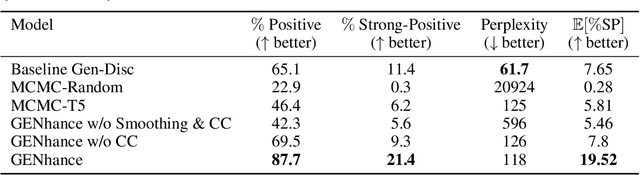
Attribute extrapolation in sample generation is challenging for deep neural networks operating beyond the training distribution. We formulate a new task for extrapolation in sequence generation, focusing on natural language and proteins, and propose GENhance, a generative framework that enhances attributes through a learned latent space. Trained on movie reviews and a computed protein stability dataset, GENhance can generate strongly-positive text reviews and highly stable protein sequences without being exposed to similar data during training. We release our benchmark tasks and models to contribute to the study of generative modeling extrapolation and data-driven design in biology and chemistry.
Profile Prediction: An Alignment-Based Pre-Training Task for Protein Sequence Models
Dec 01, 2020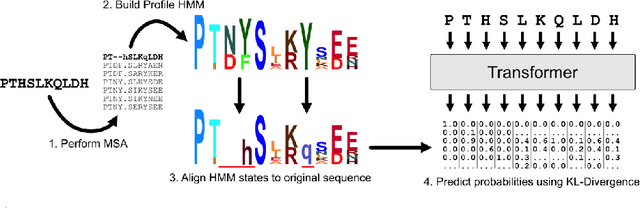
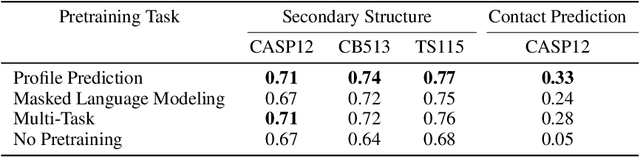
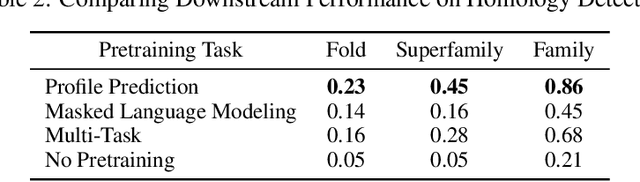
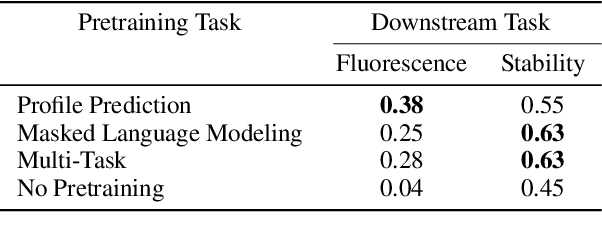
For protein sequence datasets, unlabeled data has greatly outpaced labeled data due to the high cost of wet-lab characterization. Recent deep-learning approaches to protein prediction have shown that pre-training on unlabeled data can yield useful representations for downstream tasks. However, the optimal pre-training strategy remains an open question. Instead of strictly borrowing from natural language processing (NLP) in the form of masked or autoregressive language modeling, we introduce a new pre-training task: directly predicting protein profiles derived from multiple sequence alignments. Using a set of five, standardized downstream tasks for protein models, we demonstrate that our pre-training task along with a multi-task objective outperforms masked language modeling alone on all five tasks. Our results suggest that protein sequence models may benefit from leveraging biologically-inspired inductive biases that go beyond existing language modeling techniques in NLP.
BERTology Meets Biology: Interpreting Attention in Protein Language Models
Jul 13, 2020Transformer architectures have proven to learn useful representations for protein classification and generation tasks. However, these representations present challenges in interpretability. Through the lens of attention, we analyze the inner workings of the Transformer and explore how the model discerns structural and functional properties of proteins. We show that attention (1) captures the folding structure of proteins, connecting amino acids that are far apart in the underlying sequence, but spatially close in the three-dimensional structure, (2) targets binding sites, a key functional component of proteins, and (3) focuses on progressively more complex biophysical properties with increasing layer depth. We also present a three-dimensional visualization of the interaction between attention and protein structure. Our findings align with known biological processes and provide a tool to aid discovery in protein engineering and synthetic biology. The code for visualization and analysis is available at https://github.com/salesforce/provis.
ProGen: Language Modeling for Protein Generation
Mar 08, 2020Generative modeling for protein engineering is key to solving fundamental problems in synthetic biology, medicine, and material science. We pose protein engineering as an unsupervised sequence generation problem in order to leverage the exponentially growing set of proteins that lack costly, structural annotations. We train a 1.2B-parameter language model, ProGen, on ~280M protein sequences conditioned on taxonomic and keyword tags such as molecular function and cellular component. This provides ProGen with an unprecedented range of evolutionary sequence diversity and allows it to generate with fine-grained control as demonstrated by metrics based on primary sequence similarity, secondary structure accuracy, and conformational energy.
ProDyn0: Inferring calponin homology domain stretching behavior using graph neural networks
Oct 22, 2019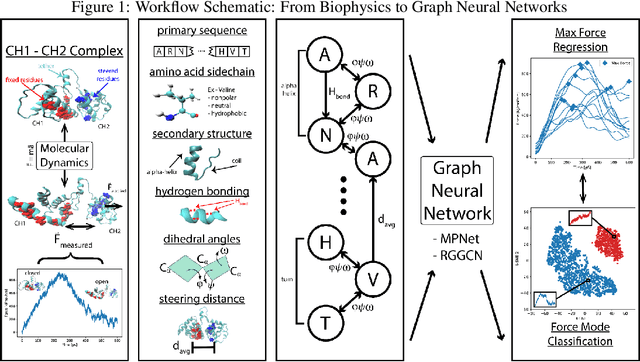
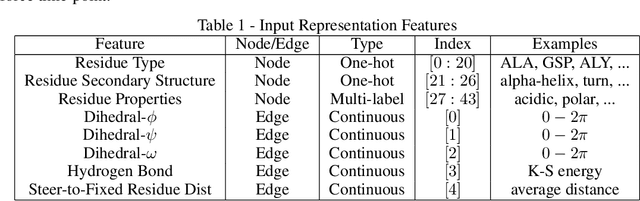
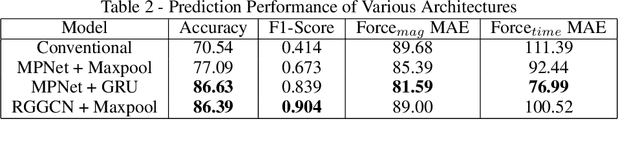
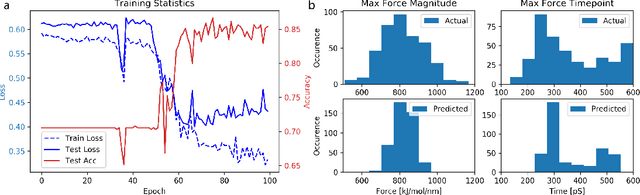
Graph neural networks are a quickly emerging field for non-Euclidean data that leverage the inherent graphical structure to predict node, edge, and global-level properties of a system. Protein properties can not easily be understood as a simple sum of their parts (i.e. amino acids), therefore, understanding their dynamical properties in the context of graphs is attractive for revealing how perturbations to their structure can affect their global function. To tackle this problem, we generate a database of 2020 mutated calponin homology (CH) domains undergoing large-scale separation in molecular dynamics. To predict the mechanosensitive force response, we develop neural message passing networks and residual gated graph convnets which predict the protein dependent force separation at 86.63 percent, 81.59 kJ/mol/nm MAE, 76.99 psec MAE for force mode classification, max force magnitude, max force time respectively-- significantly better than non-graph-based deep learning techniques. Towards uniting geometric learning techniques and biophysical observables, we premiere our simulation database as a benchmark dataset for further development/evaluation of graph neural network architectures.
* 8 pages, 2 figures, 2 tables
Bimodal network architectures for automatic generation of image annotation from text
Sep 05, 2018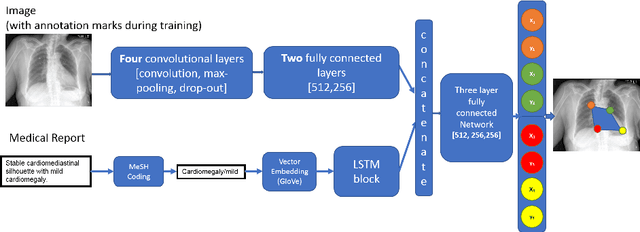

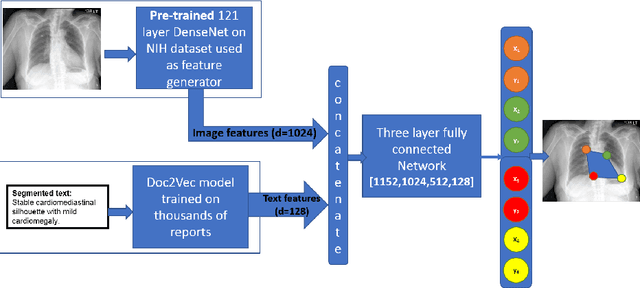
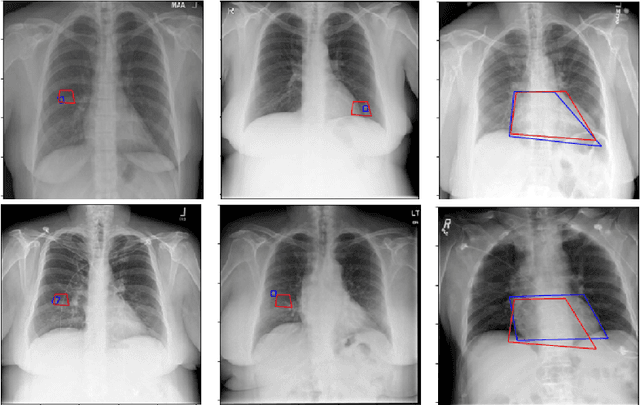
Medical image analysis practitioners have embraced big data methodologies. This has created a need for large annotated datasets. The source of big data is typically large image collections and clinical reports recorded for these images. In many cases, however, building algorithms aimed at segmentation and detection of disease requires a training dataset with markings of the areas of interest on the image that match with the described anomalies. This process of annotation is expensive and needs the involvement of clinicians. In this work we propose two separate deep neural network architectures for automatic marking of a region of interest (ROI) on the image best representing a finding location, given a textual report or a set of keywords. One architecture consists of LSTM and CNN components and is trained end to end with images, matching text, and markings of ROIs for those images. The output layer estimates the coordinates of the vertices of a polygonal region. The second architecture uses a network pre-trained on a large dataset of the same image types for learning feature representations of the findings of interest. We show that for a variety of findings from chest X-ray images, both proposed architectures learn to estimate the ROI, as validated by clinical annotations. There is a clear advantage obtained from the architecture with pre-trained imaging network. The centroids of the ROIs marked by this network were on average at a distance equivalent to 5.1% of the image width from the centroids of the ground truth ROIs.
* Accepted to MICCAI 2018, LNCS 11070
Fast and accurate classification of echocardiograms using deep learning
Jun 27, 2017
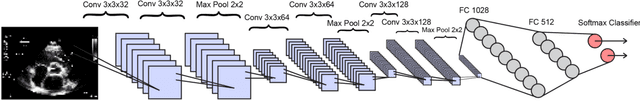
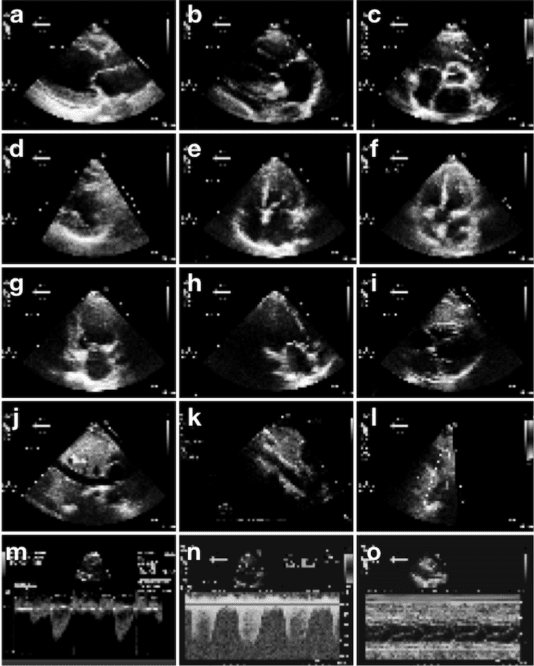
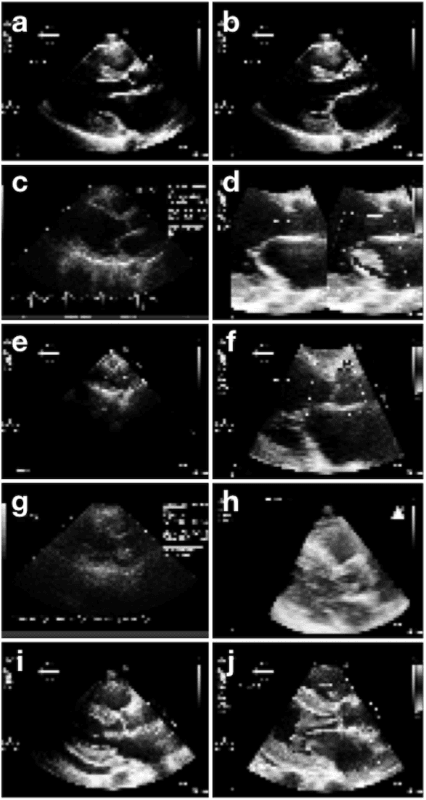
Echocardiography is essential to modern cardiology. However, human interpretation limits high throughput analysis, limiting echocardiography from reaching its full clinical and research potential for precision medicine. Deep learning is a cutting-edge machine-learning technique that has been useful in analyzing medical images but has not yet been widely applied to echocardiography, partly due to the complexity of echocardiograms' multi view, multi modality format. The essential first step toward comprehensive computer assisted echocardiographic interpretation is determining whether computers can learn to recognize standard views. To this end, we anonymized 834,267 transthoracic echocardiogram (TTE) images from 267 patients (20 to 96 years, 51 percent female, 26 percent obese) seen between 2000 and 2017 and labeled them according to standard views. Images covered a range of real world clinical variation. We built a multilayer convolutional neural network and used supervised learning to simultaneously classify 15 standard views. Eighty percent of data used was randomly chosen for training and 20 percent reserved for validation and testing on never seen echocardiograms. Using multiple images from each clip, the model classified among 12 video views with 97.8 percent overall test accuracy without overfitting. Even on single low resolution images, test accuracy among 15 views was 91.7 percent versus 70.2 to 83.5 percent for board-certified echocardiographers. Confusional matrices, occlusion experiments, and saliency mapping showed that the model finds recognizable similarities among related views and classifies using clinically relevant image features. In conclusion, deep neural networks can classify essential echocardiographic views simultaneously and with high accuracy. Our results provide a foundation for more complex deep learning assisted echocardiographic interpretation.