 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBayesian Empirical Bayes: Simultaneous Inference from Probabilistic Symmetries
Dec 22, 2025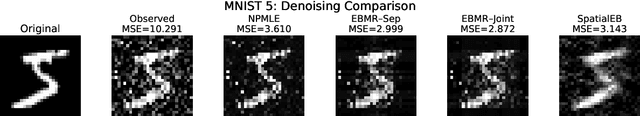
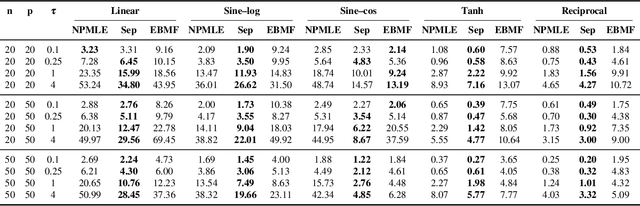
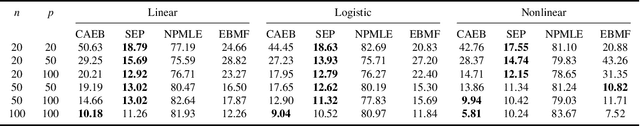
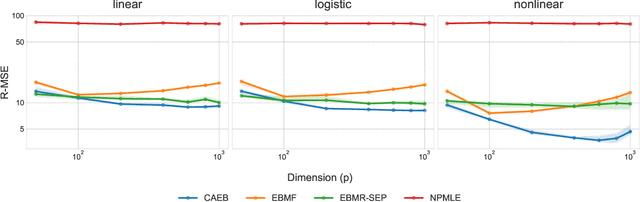
Empirical Bayes (EB) improves the accuracy of simultaneous inference "by learning from the experience of others" (Efron, 2012). Classical EB theory focuses on latent variables that are iid draws from a fitted prior (Efron, 2019). Modern applications, however, feature complex structure, like arrays, spatial processes, or covariates. How can we apply EB ideas to these settings? We propose a generalized approach to empirical Bayes based on the notion of probabilistic symmetry. Our method pairs a simultaneous inference problem-with an unknown prior-to a symmetry assumption on the joint distribution of the latent variables. Each symmetry implies an ergodic decomposition, which we use to derive a corresponding empirical Bayes method. We call this methodBayesian empirical Bayes (BEB). We show how BEB recovers the classical methods of empirical Bayes, which implicitly assume exchangeability. We then use it to extend EB to other probabilistic symmetries: (i) EB matrix recovery for arrays and graphs; (ii) covariate-assisted EB for conditional data; (iii) EB spatial regression under shift invariance. We develop scalable algorithms based on variational inference and neural networks. In simulations, BEB outperforms existing approaches to denoising arrays and spatial data. On real data, we demonstrate BEB by denoising a cancer gene-expression matrix and analyzing spatial air-quality data from New York City.
Lifting Biomolecular Data Acquisition
Dec 17, 2025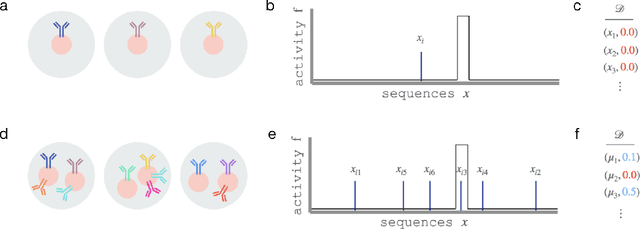
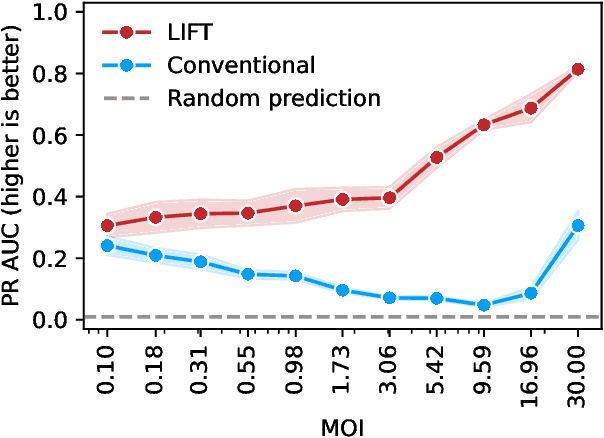
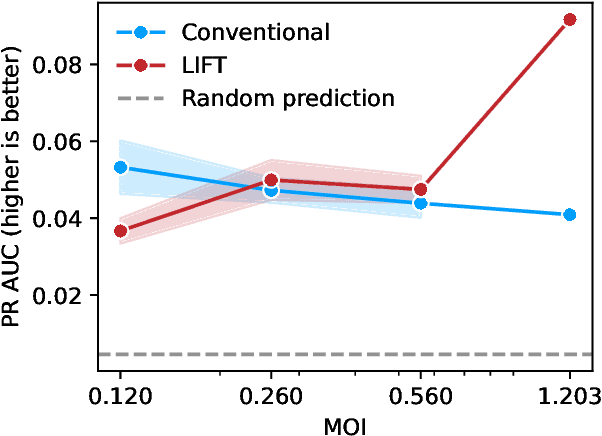
One strategy to scale up ML-driven science is to increase wet lab experiments' information density. We present a method based on a neural extension of compressed sensing to function space. We measure the activity of multiple different molecules simultaneously, rather than individually. Then, we deconvolute the molecule-activity map during model training. Co-design of wet lab experiments and learning algorithms provably leads to orders-of-magnitude gains in information density. We demonstrate on antibodies and cell therapies.
Adaptive Nonparametric Perturbations of Parametric Bayesian Models
Dec 17, 2024



Parametric Bayesian modeling offers a powerful and flexible toolbox for scientific data analysis. Yet the model, however detailed, may still be wrong, and this can make inferences untrustworthy. In this paper we study nonparametrically perturbed parametric (NPP) Bayesian models, in which a parametric Bayesian model is relaxed via a distortion of its likelihood. We analyze the properties of NPP models when the target of inference is the true data distribution or some functional of it, such as in causal inference. We show that NPP models can offer the robustness of nonparametric models while retaining the data efficiency of parametric models, achieving fast convergence when the parametric model is close to true. To efficiently analyze data with an NPP model, we develop a generalized Bayes procedure to approximate its posterior. We demonstrate our method by estimating causal effects of gene expression from single cell RNA sequencing data. NPP modeling offers an efficient approach to robust Bayesian inference and can be used to robustify any parametric Bayesian model.
Estimating the Causal Effects of T Cell Receptors
Oct 18, 2024



A central question in human immunology is how a patient's repertoire of T cells impacts disease. Here, we introduce a method to infer the causal effects of T cell receptor (TCR) sequences on patient outcomes using observational TCR repertoire sequencing data and clinical outcomes data. Our approach corrects for unobserved confounders, such as a patient's environment and life history, by using the patient's immature, pre-selection TCR repertoire. The pre-selection repertoire can be estimated from nonproductive TCR data, which is widely available. It is generated by a randomized mutational process, V(D)J recombination, which provides a natural experiment. We show formally how to use the pre-selection repertoire to draw causal inferences, and develop a scalable neural-network estimator for our identification formula. Our method produces an estimate of the effect of interventions that add a specific TCR sequence to patient repertoires. As a demonstration, we use it to analyze the effects of TCRs on COVID-19 severity, uncovering potentially therapeutic TCRs that are (1) observed in patients, (2) bind SARS-CoV-2 antigens in vitro and (3) have strong positive effects on clinical outcomes.
Hierarchical Causal Models
Jan 10, 2024Scientists often want to learn about cause and effect from hierarchical data, collected from subunits nested inside units. Consider students in schools, cells in patients, or cities in states. In such settings, unit-level variables (e.g. each school's budget) may affect subunit-level variables (e.g. the test scores of each student in each school) and vice versa. To address causal questions with hierarchical data, we propose hierarchical causal models, which extend structural causal models and causal graphical models by adding inner plates. We develop a general graphical identification technique for hierarchical causal models that extends do-calculus. We find many situations in which hierarchical data can enable causal identification even when it would be impossible with non-hierarchical data, that is, if we had only unit-level summaries of subunit-level variables (e.g. the school's average test score, rather than each student's score). We develop estimation techniques for hierarchical causal models, using methods including hierarchical Bayesian models. We illustrate our results in simulation and via a reanalysis of the classic "eight schools" study.
ProGen2: Exploring the Boundaries of Protein Language Models
Jun 27, 2022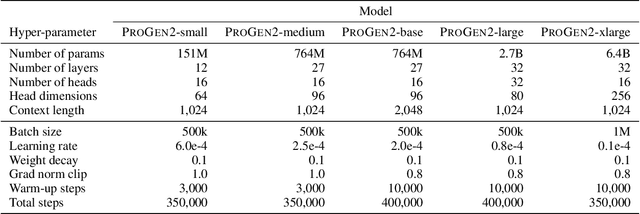
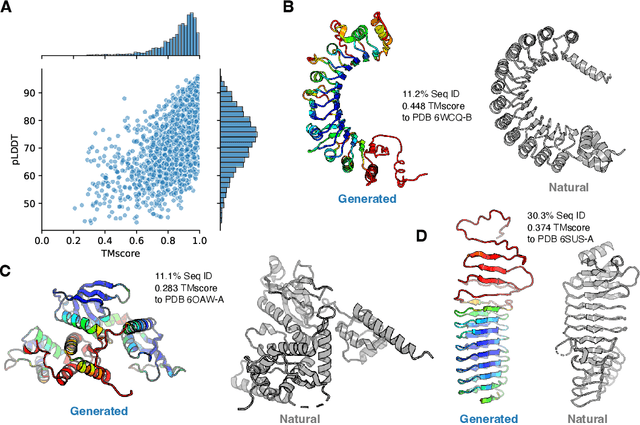
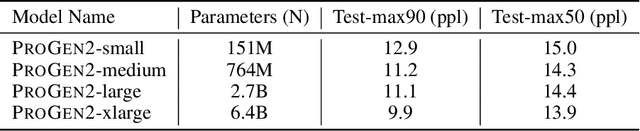
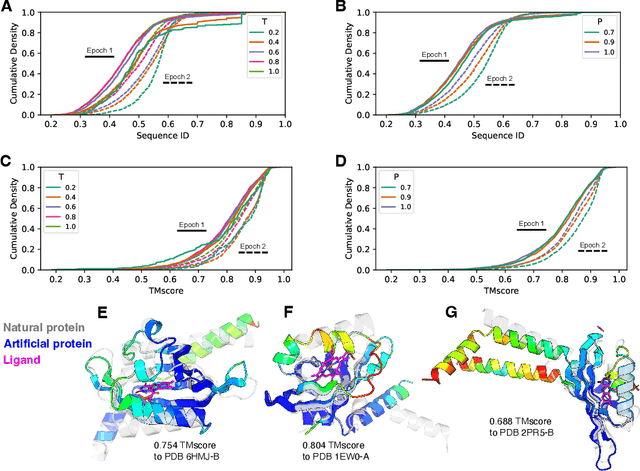
Attention-based models trained on protein sequences have demonstrated incredible success at classification and generation tasks relevant for artificial intelligence-driven protein design. However, we lack a sufficient understanding of how very large-scale models and data play a role in effective protein model development. We introduce a suite of protein language models, named ProGen2, that are scaled up to 6.4B parameters and trained on different sequence datasets drawn from over a billion proteins from genomic, metagenomic, and immune repertoire databases. ProGen2 models show state-of-the-art performance in capturing the distribution of observed evolutionary sequences, generating novel viable sequences, and predicting protein fitness without additional finetuning. As large model sizes and raw numbers of protein sequences continue to become more widely accessible, our results suggest that a growing emphasis needs to be placed on the data distribution provided to a protein sequence model. We release the ProGen2 models and code at https://github.com/salesforce/progen.