 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTopotein: Topological Deep Learning for Protein Representation Learning
Sep 04, 2025Protein representation learning (PRL) is crucial for understanding structure-function relationships, yet current sequence- and graph-based methods fail to capture the hierarchical organization inherent in protein structures. We introduce Topotein, a comprehensive framework that applies topological deep learning to PRL through the novel Protein Combinatorial Complex (PCC) and Topology-Complete Perceptron Network (TCPNet). Our PCC represents proteins at multiple hierarchical levels -- from residues to secondary structures to complete proteins -- while preserving geometric information at each level. TCPNet employs SE(3)-equivariant message passing across these hierarchical structures, enabling more effective capture of multi-scale structural patterns. Through extensive experiments on four PRL tasks, TCPNet consistently outperforms state-of-the-art geometric graph neural networks. Our approach demonstrates particular strength in tasks such as fold classification which require understanding of secondary structure arrangements, validating the importance of hierarchical topological features for protein analysis.
Structure-based drug design by denoising voxel grids
May 07, 2024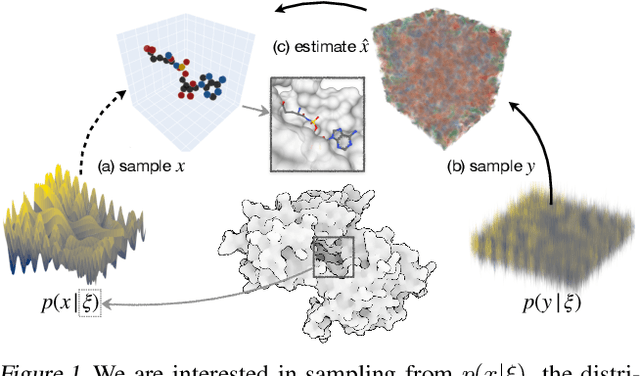
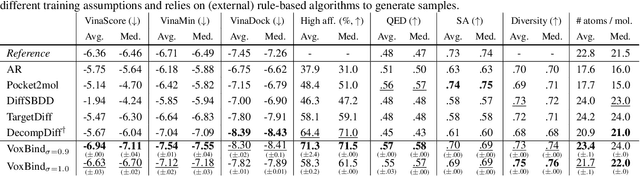
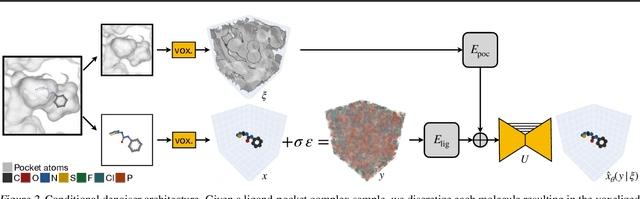
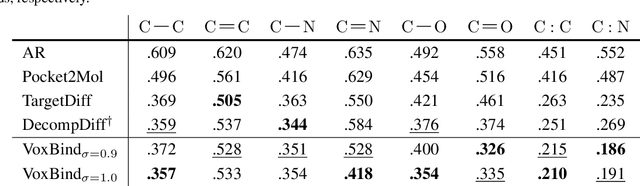
We present VoxBind, a new score-based generative model for 3D molecules conditioned on protein structures. Our approach represents molecules as 3D atomic density grids and leverages a 3D voxel-denoising network for learning and generation. We extend the neural empirical Bayes formalism (Saremi & Hyvarinen, 2019) to the conditional setting and generate structure-conditioned molecules with a two-step procedure: (i) sample noisy molecules from the Gaussian-smoothed conditional distribution with underdamped Langevin MCMC using the learned score function and (ii) estimate clean molecules from the noisy samples with single-step denoising. Compared to the current state of the art, our model is simpler to train, significantly faster to sample from, and achieves better results on extensive in silico benchmarks -- the generated molecules are more diverse, exhibit fewer steric clashes, and bind with higher affinity to protein pockets.
GAUCHE: A Library for Gaussian Processes in Chemistry
Dec 06, 2022We introduce GAUCHE, a library for GAUssian processes in CHEmistry. Gaussian processes have long been a cornerstone of probabilistic machine learning, affording particular advantages for uncertainty quantification and Bayesian optimisation. Extending Gaussian processes to chemical representations, however, is nontrivial, necessitating kernels defined over structured inputs such as graphs, strings and bit vectors. By defining such kernels in GAUCHE, we seek to open the door to powerful tools for uncertainty quantification and Bayesian optimisation in chemistry. Motivated by scenarios frequently encountered in experimental chemistry, we showcase applications for GAUCHE in molecular discovery and chemical reaction optimisation. The codebase is made available at https://github.com/leojklarner/gauche
Structure-based Drug Design with Equivariant Diffusion Models
Oct 24, 2022Structure-based drug design (SBDD) aims to design small-molecule ligands that bind with high affinity and specificity to pre-determined protein targets. Traditional SBDD pipelines start with large-scale docking of compound libraries from public databases, thus limiting the exploration of chemical space to existent previously studied regions. Recent machine learning methods approached this problem using an atom-by-atom generation approach, which is computationally expensive. In this paper, we formulate SBDD as a 3D-conditional generation problem and present DiffSBDD, an E(3)-equivariant 3D-conditional diffusion model that generates novel ligands conditioned on protein pockets. Furthermore, we curate a new dataset of experimentally determined binding complex data from Binding MOAD to provide a realistic binding scenario that complements the synthetic CrossDocked dataset. Comprehensive in silico experiments demonstrate the efficiency of DiffSBDD in generating novel and diverse drug-like ligands that engage protein pockets with high binding energies as predicted by in silico docking.
Protein Representation Learning by Geometric Structure Pretraining
Mar 14, 2022
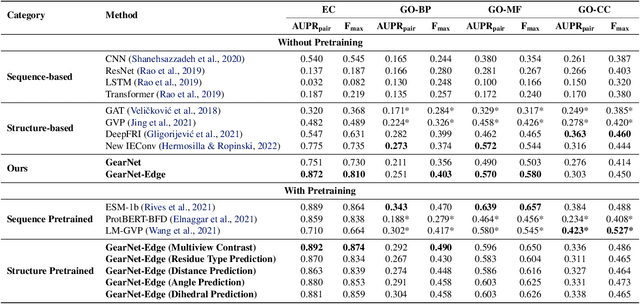

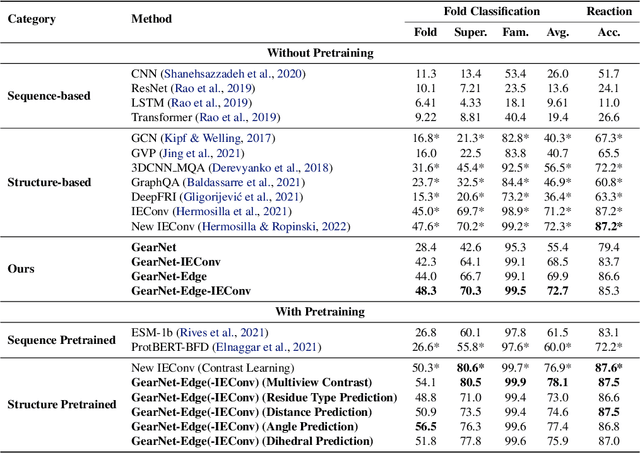
Learning effective protein representations is critical in a variety of tasks in biology such as predicting protein function or structure. Existing approaches usually pretrain protein language models on a large number of unlabeled amino acid sequences and then finetune the models with some labeled data in downstream tasks. Despite the effectiveness of sequence-based approaches, the power of pretraining on smaller numbers of known protein structures has not been explored for protein property prediction, though protein structures are known to be determinants of protein function. We first present a simple yet effective encoder to learn protein geometry features. We pretrain the protein graph encoder by leveraging multiview contrastive learning and different self-prediction tasks. Experimental results on both function prediction and fold classification tasks show that our proposed pretraining methods outperform or are on par with the state-of-the-art sequence-based methods using much less data. All codes and models will be published upon acceptance.