 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNEBULA: Neural Empirical Bayes Under Latent Representations for Efficient and Controllable Design of Molecular Libraries
Jul 03, 2024We present NEBULA, the first latent 3D generative model for scalable generation of large molecular libraries around a seed compound of interest. Such libraries are crucial for scientific discovery, but it remains challenging to generate large numbers of high quality samples efficiently. 3D-voxel-based methods have recently shown great promise for generating high quality samples de novo from random noise (Pinheiro et al., 2023). However, sampling in 3D-voxel space is computationally expensive and use in library generation is prohibitively slow. Here, we instead perform neural empirical Bayes sampling (Saremi & Hyvarinen, 2019) in the learned latent space of a vector-quantized variational autoencoder. NEBULA generates large molecular libraries nearly an order of magnitude faster than existing methods without sacrificing sample quality. Moreover, NEBULA generalizes better to unseen drug-like molecules, as demonstrated on two public datasets and multiple recently released drugs. We expect the approach herein to be highly enabling for machine learning-based drug discovery. The code is available at https://github.com/prescient-design/nebula
Structure-based drug design by denoising voxel grids
May 07, 2024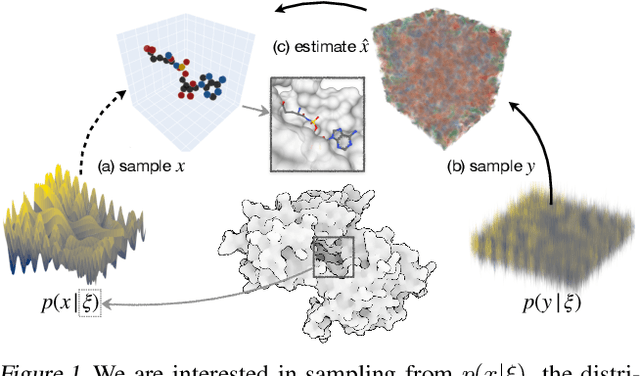
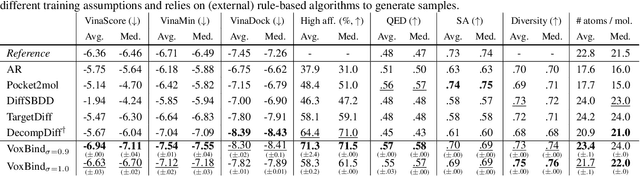
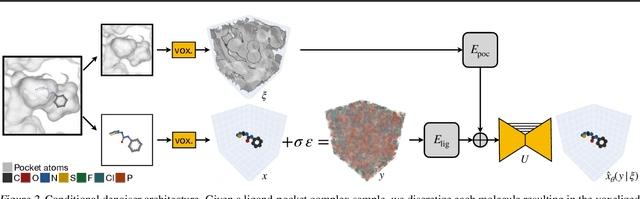
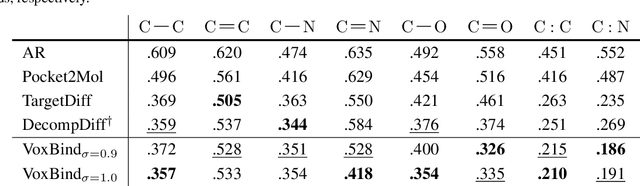
We present VoxBind, a new score-based generative model for 3D molecules conditioned on protein structures. Our approach represents molecules as 3D atomic density grids and leverages a 3D voxel-denoising network for learning and generation. We extend the neural empirical Bayes formalism (Saremi & Hyvarinen, 2019) to the conditional setting and generate structure-conditioned molecules with a two-step procedure: (i) sample noisy molecules from the Gaussian-smoothed conditional distribution with underdamped Langevin MCMC using the learned score function and (ii) estimate clean molecules from the noisy samples with single-step denoising. Compared to the current state of the art, our model is simpler to train, significantly faster to sample from, and achieves better results on extensive in silico benchmarks -- the generated molecules are more diverse, exhibit fewer steric clashes, and bind with higher affinity to protein pockets.
3D molecule generation by denoising voxel grids
Jun 13, 2023We propose a new score-based approach to generate 3D molecules represented as atomic densities on regular grids. First, we train a denoising neural network that learns to map from a smooth distribution of noisy molecules to the distribution of real molecules. Then, we follow the neural empirical Bayes framework [Saremi and Hyvarinen, 2019] and generate molecules in two steps: (i) sample noisy density grids from a smooth distribution via underdamped Langevin Markov chain Monte Carlo, and (ii) recover the ``clean'' molecule by denoising the noisy grid with a single step. Our method, VoxMol, generates molecules in a fundamentally different way than the current state of the art (i.e., diffusion models applied to atom point clouds). It differs in terms of the data representation, the noise model, the network architecture and the generative modeling algorithm. VoxMol achieves comparable results to state of the art on unconditional 3D molecule generation while being simpler to train and faster to generate molecules.
A COLD Approach to Generating Optimal Samples
May 23, 2019
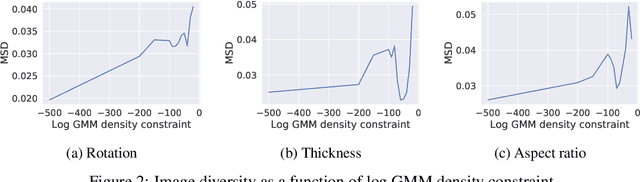
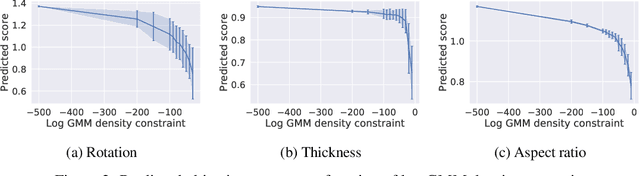
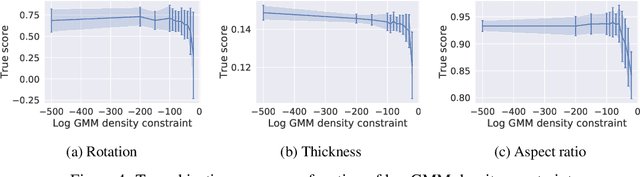
Optimising discrete data for a desired characteristic using gradient-based methods involves projecting the data into a continuous latent space and carrying out optimisation in this space. Carrying out global optimisation is difficult as optimisers are likely to follow gradients into regions of the latent space that the model has not been exposed to during training; samples generated from these regions are likely to be too dissimilar to the training data to be useful. We propose Constrained Optimisation with Latent Distributions (COLD), a constrained global optimisation procedure to find samples with high values of a desired property that are similar to yet distinct from the training data. We find that on MNIST, our procedure yields optima for each of three different objectives, and that enforcing tighter constraints improves the quality and increases the diversity of the generated images. On the ChEMBL molecular dataset, our method generates a diverse set of new molecules with drug-likeness scores similar to those of the highest-scoring molecules in the training data. We also demonstrate a computationally efficient way to approximate the constraint when evaluating it exactly is computationally expensive.
Molecular geometry prediction using a deep generative graph neural network
Mar 31, 2019
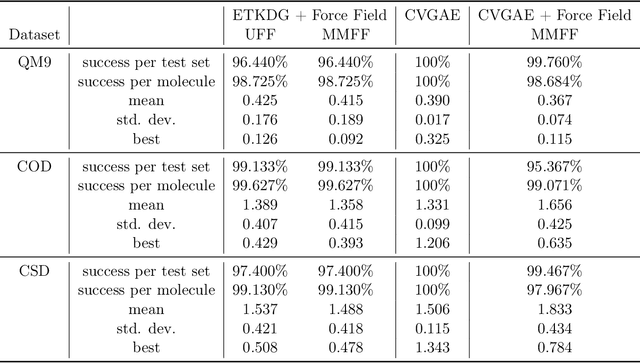
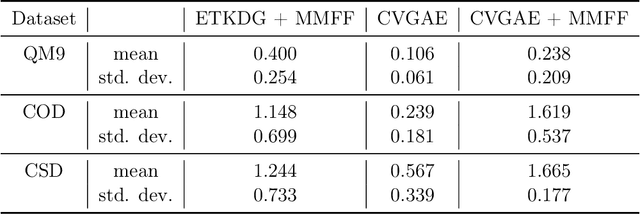
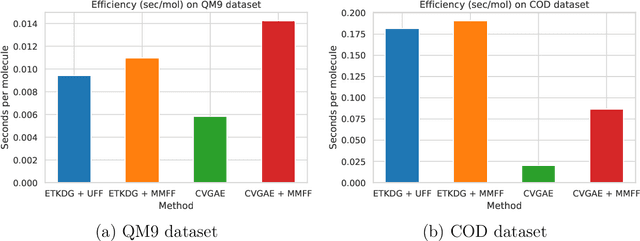
A molecule's geometry, also known as conformation, is one of a molecule's most important properties, determining the reactions it participates in, the bonds it forms, and the interactions it has with other molecules. Conventional conformation generation methods minimize hand-designed molecular force field energy functions that are not well correlated with the true energy function of a molecule observed in nature. They generate geometrically diverse sets of conformations, some of which are very similar to the ground-truth conformations and others of which are very different. In this paper we propose a conditional deep generative graph neural network that learns an energy function from data by directly learning to generate molecular conformations given a molecular graph. On three large scale small molecule datasets, we show that our method generates a set of conformations that on average is far more likely to be close to the corresponding reference conformations than are those obtained from conventional force field methods. Our method maintains geometrical diversity by generating conformations that are not too similar to each other, and is also computationally faster. We also show that our method can be used to provide initial coordinates for conventional force field methods. On one of the evaluated datasets we show that this combination allows us to combine the best of both methods, yielding generated conformations that are on average close to ground-truth conformations with some very similar to ground-truth conformations.