 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecular geometry prediction using a deep generative graph neural network
Paper and Code
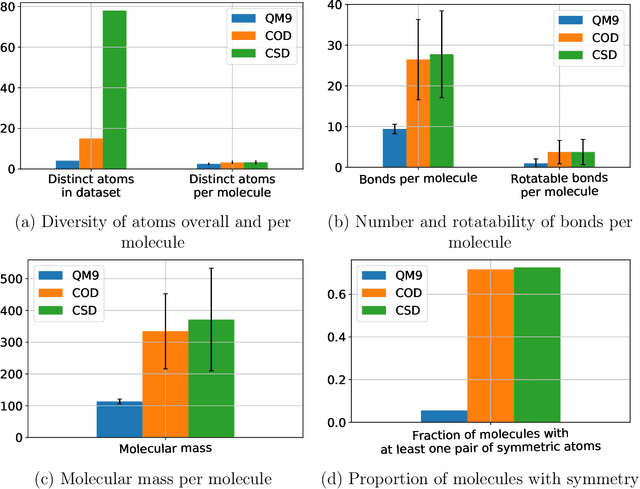
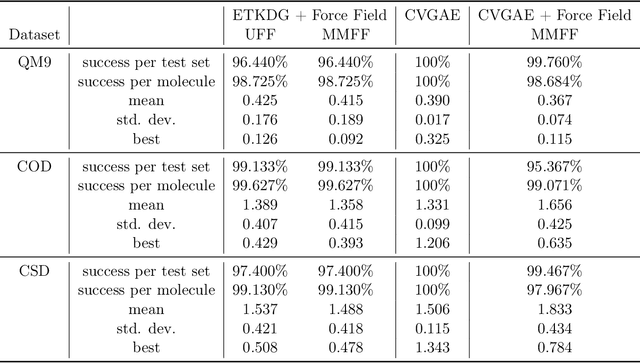
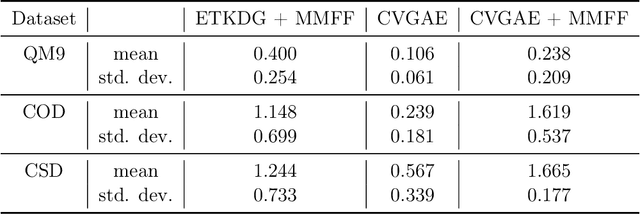
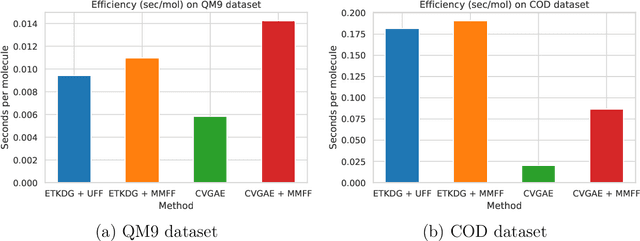
A molecule's geometry, also known as conformation, is one of a molecule's most important properties, determining the reactions it participates in, the bonds it forms, and the interactions it has with other molecules. Conventional conformation generation methods minimize hand-designed molecular force field energy functions that are not well correlated with the true energy function of a molecule observed in nature. They generate geometrically diverse sets of conformations, some of which are very similar to the ground-truth conformations and others of which are very different. In this paper we propose a conditional deep generative graph neural network that learns an energy function from data by directly learning to generate molecular conformations given a molecular graph. On three large scale small molecule datasets, we show that our method generates a set of conformations that on average is far more likely to be close to the corresponding reference conformations than are those obtained from conventional force field methods. Our method maintains geometrical diversity by generating conformations that are not too similar to each other, and is also computationally faster. We also show that our method can be used to provide initial coordinates for conventional force field methods. On one of the evaluated datasets we show that this combination allows us to combine the best of both methods, yielding generated conformations that are on average close to ground-truth conformations with some very similar to ground-truth conformations.