 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePaAno: Patch-Based Representation Learning for Time-Series Anomaly Detection
Feb 01, 2026Although recent studies on time-series anomaly detection have increasingly adopted ever-larger neural network architectures such as transformers and foundation models, they incur high computational costs and memory usage, making them impractical for real-time and resource-constrained scenarios. Moreover, they often fail to demonstrate significant performance gains over simpler methods under rigorous evaluation protocols. In this study, we propose Patch-based representation learning for time-series Anomaly detection (PaAno), a lightweight yet effective method for fast and efficient time-series anomaly detection. PaAno extracts short temporal patches from time-series training data and uses a 1D convolutional neural network to embed each patch into a vector representation. The model is trained using a combination of triplet loss and pretext loss to ensure the embeddings capture informative temporal patterns from input patches. During inference, the anomaly score at each time step is computed by comparing the embeddings of its surrounding patches to those of normal patches extracted from the training time-series. Evaluated on the TSB-AD benchmark, PaAno achieved state-of-the-art performance, significantly outperforming existing methods, including those based on heavy architectures, on both univariate and multivariate time-series anomaly detection across various range-wise and point-wise performance measures.
AnoStyler: Text-Driven Localized Anomaly Generation via Lightweight Style Transfer
Nov 10, 2025Anomaly generation has been widely explored to address the scarcity of anomaly images in real-world data. However, existing methods typically suffer from at least one of the following limitations, hindering their practical deployment: (1) lack of visual realism in generated anomalies; (2) dependence on large amounts of real images; and (3) use of memory-intensive, heavyweight model architectures. To overcome these limitations, we propose AnoStyler, a lightweight yet effective method that frames zero-shot anomaly generation as text-guided style transfer. Given a single normal image along with its category label and expected defect type, an anomaly mask indicating the localized anomaly regions and two-class text prompts representing the normal and anomaly states are generated using generalizable category-agnostic procedures. A lightweight U-Net model trained with CLIP-based loss functions is used to stylize the normal image into a visually realistic anomaly image, where anomalies are localized by the anomaly mask and semantically aligned with the text prompts. Extensive experiments on the MVTec-AD and VisA datasets show that AnoStyler outperforms existing anomaly generation methods in generating high-quality and diverse anomaly images. Furthermore, using these generated anomalies helps enhance anomaly detection performance.
Molecular geometry prediction using a deep generative graph neural network
Mar 31, 2019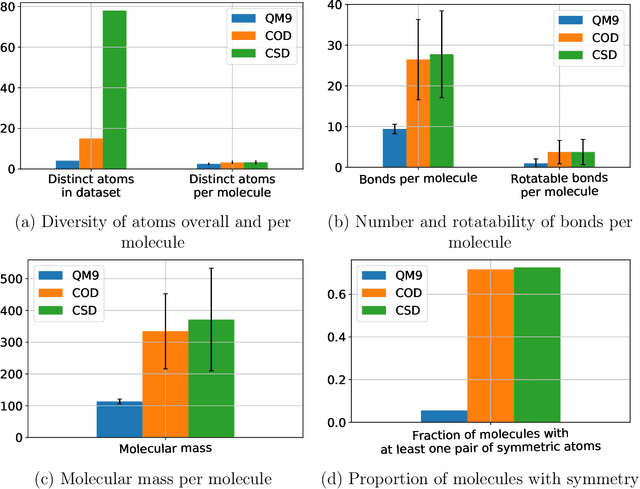
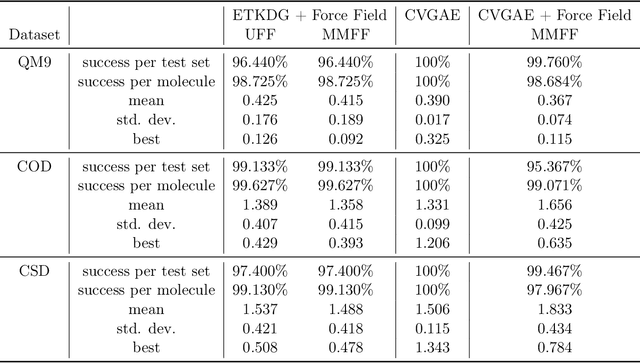
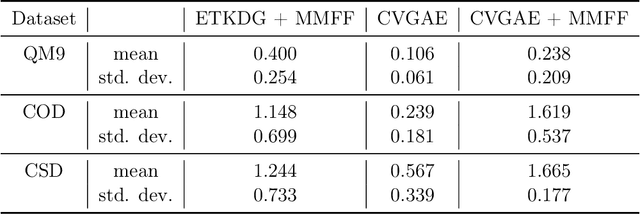
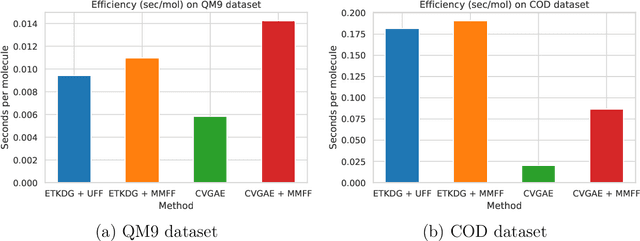
A molecule's geometry, also known as conformation, is one of a molecule's most important properties, determining the reactions it participates in, the bonds it forms, and the interactions it has with other molecules. Conventional conformation generation methods minimize hand-designed molecular force field energy functions that are not well correlated with the true energy function of a molecule observed in nature. They generate geometrically diverse sets of conformations, some of which are very similar to the ground-truth conformations and others of which are very different. In this paper we propose a conditional deep generative graph neural network that learns an energy function from data by directly learning to generate molecular conformations given a molecular graph. On three large scale small molecule datasets, we show that our method generates a set of conformations that on average is far more likely to be close to the corresponding reference conformations than are those obtained from conventional force field methods. Our method maintains geometrical diversity by generating conformations that are not too similar to each other, and is also computationally faster. We also show that our method can be used to provide initial coordinates for conventional force field methods. On one of the evaluated datasets we show that this combination allows us to combine the best of both methods, yielding generated conformations that are on average close to ground-truth conformations with some very similar to ground-truth conformations.
Conditional molecular design with deep generative models
Jul 23, 2018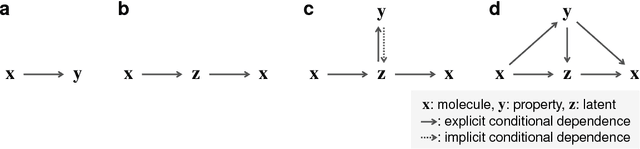
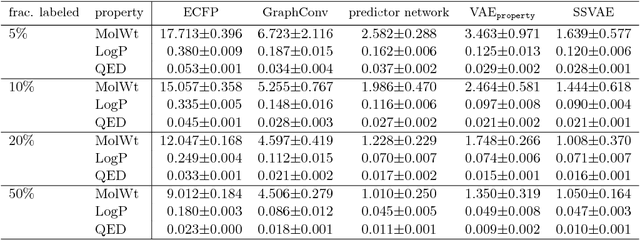
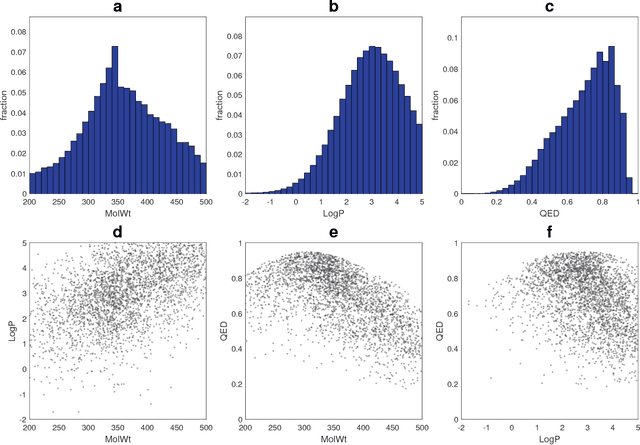
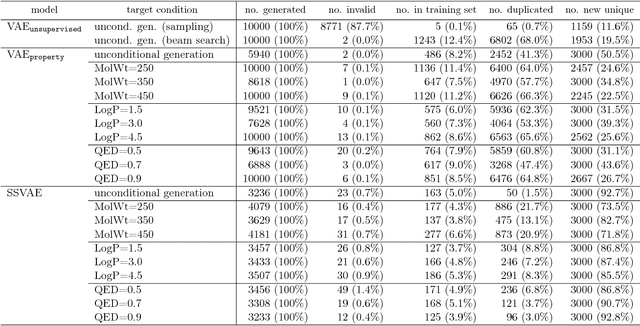
Although machine learning has been successfully used to propose novel molecules that satisfy desired properties, it is still challenging to explore a large chemical space efficiently. In this paper, we present a conditional molecular design method that facilitates generating new molecules with desired properties. The proposed model, which simultaneously performs both property prediction and molecule generation, is built as a semi-supervised variational autoencoder trained on a set of existing molecules with only a partial annotation. We generate new molecules with desired properties by sampling from the generative distribution estimated by the model. We demonstrate the effectiveness of the proposed model by evaluating it on drug-like molecules. The model improves the performance of property prediction by exploiting unlabeled molecules, and efficiently generates novel molecules fulfilling various target conditions.