 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSimilarity-Quantized Relative Difference Learning for Improved Molecular Activity Prediction
Jan 15, 2025



Accurate prediction of molecular activities is crucial for efficient drug discovery, yet remains challenging due to limited and noisy datasets. We introduce Similarity-Quantized Relative Learning (SQRL), a learning framework that reformulates molecular activity prediction as relative difference learning between structurally similar pairs of compounds. SQRL uses precomputed molecular similarities to enhance training of graph neural networks and other architectures, and significantly improves accuracy and generalization in low-data regimes common in drug discovery. We demonstrate its broad applicability and real-world potential through benchmarking on public datasets as well as proprietary industry data. Our findings demonstrate that leveraging similarity-aware relative differences provides an effective paradigm for molecular activity prediction.
Structure-based drug design by denoising voxel grids
May 07, 2024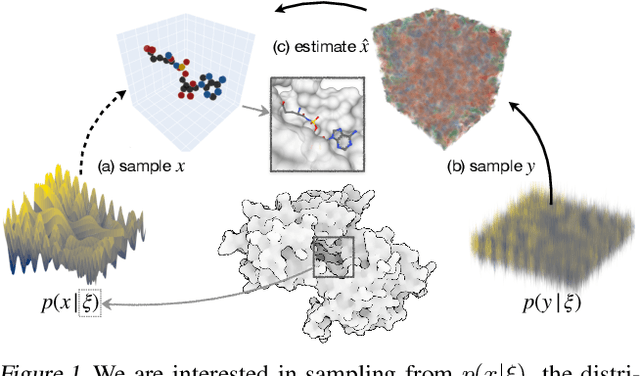
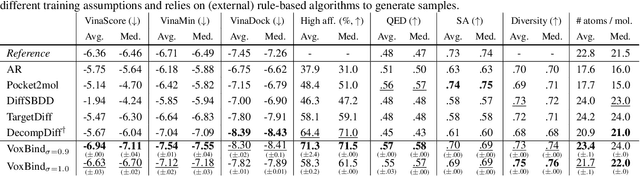
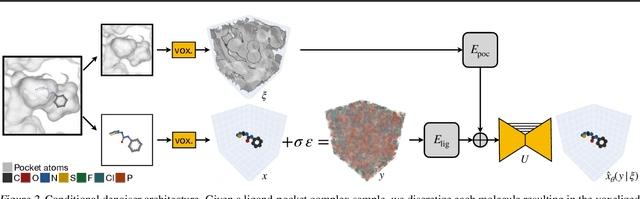
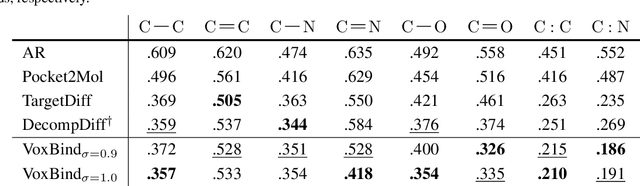
We present VoxBind, a new score-based generative model for 3D molecules conditioned on protein structures. Our approach represents molecules as 3D atomic density grids and leverages a 3D voxel-denoising network for learning and generation. We extend the neural empirical Bayes formalism (Saremi & Hyvarinen, 2019) to the conditional setting and generate structure-conditioned molecules with a two-step procedure: (i) sample noisy molecules from the Gaussian-smoothed conditional distribution with underdamped Langevin MCMC using the learned score function and (ii) estimate clean molecules from the noisy samples with single-step denoising. Compared to the current state of the art, our model is simpler to train, significantly faster to sample from, and achieves better results on extensive in silico benchmarks -- the generated molecules are more diverse, exhibit fewer steric clashes, and bind with higher affinity to protein pockets.
3D molecule generation by denoising voxel grids
Jun 13, 2023We propose a new score-based approach to generate 3D molecules represented as atomic densities on regular grids. First, we train a denoising neural network that learns to map from a smooth distribution of noisy molecules to the distribution of real molecules. Then, we follow the neural empirical Bayes framework [Saremi and Hyvarinen, 2019] and generate molecules in two steps: (i) sample noisy density grids from a smooth distribution via underdamped Langevin Markov chain Monte Carlo, and (ii) recover the ``clean'' molecule by denoising the noisy grid with a single step. Our method, VoxMol, generates molecules in a fundamentally different way than the current state of the art (i.e., diffusion models applied to atom point clouds). It differs in terms of the data representation, the noise model, the network architecture and the generative modeling algorithm. VoxMol achieves comparable results to state of the art on unconditional 3D molecule generation while being simpler to train and faster to generate molecules.
Black Box Recursive Translations for Molecular Optimization
Dec 21, 2019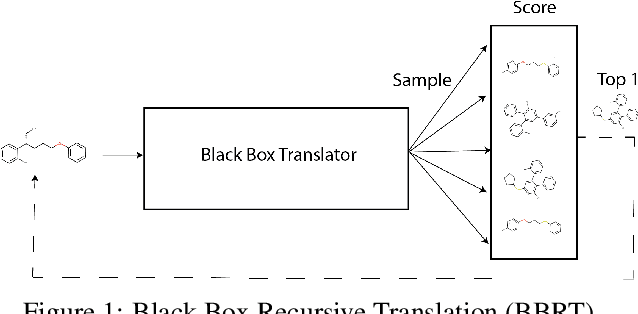
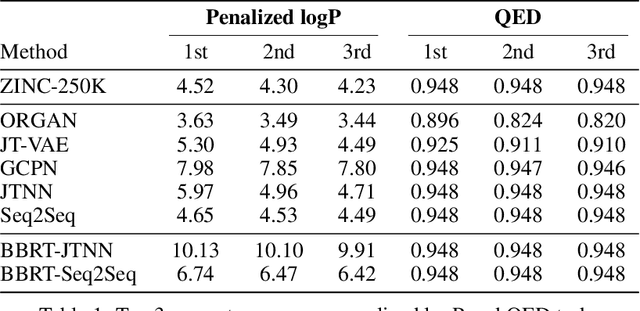
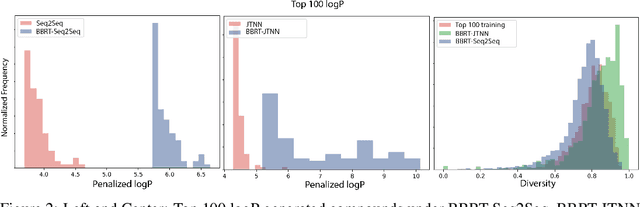
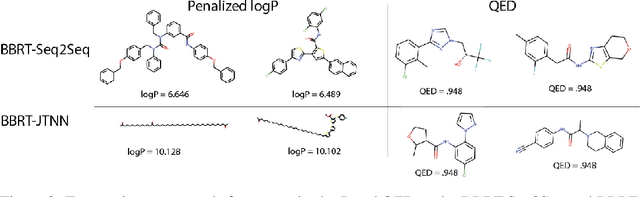
Machine learning algorithms for generating molecular structures offer a promising new approach to drug discovery. We cast molecular optimization as a translation problem, where the goal is to map an input compound to a target compound with improved biochemical properties. Remarkably, we observe that when generated molecules are iteratively fed back into the translator, molecular compound attributes improve with each step. We show that this finding is invariant to the choice of translation model, making this a "black box" algorithm. We call this method Black Box Recursive Translation (BBRT), a new inference method for molecular property optimization. This simple, powerful technique operates strictly on the inputs and outputs of any translation model. We obtain new state-of-the-art results for molecular property optimization tasks using our simple drop-in replacement with well-known sequence and graph-based models. Our method provides a significant boost in performance relative to its non-recursive peers with just a simple "for" loop. Further, BBRT is highly interpretable, allowing users to map the evolution of newly discovered compounds from known starting points.